化工进展 ›› 2019, Vol. 38 ›› Issue (07): 3253-3264.DOI: 10.16085/j.issn.1000-6613.2018-1808
密度泛函理论在高电压电解液设计中的应用
李南1( ),马国强1(),车海英2,蒋志敏1,沈旻1,董经博1,陈慧闯1,马紫峰2()
),马国强1(),车海英2,蒋志敏1,沈旻1,董经博1,陈慧闯1,马紫峰2()
- 1. 浙江省化工研究院有限公司,浙江 杭州 310023
2. 上海交通大学化学工程系,上海电化学能源器件工程技术研究中心,上海 200240
-
收稿日期:2018-09-07出版日期:2019-07-05发布日期:2019-07-05 -
通讯作者:马国强,马紫峰 -
作者简介:李南(1991—),女,硕士研究生,研究方向为锂离子电池电解液。E-mail:<email>linan15@sinochem.com</email>。 -
基金资助:国家自然科学基金重点项目(21336003)
Application of density functional theory in the design of high potential electrolyte
Nan LI1(),Guoqiang MA1(),Haiying CHE2,Zhimin JIANG1,Min SHEN1,Jingbo DONG1,Huichuang CHEN1,Zifeng MA2()
- 1. Zhejiang Research Institute of Chemical Industry Ltd. , Hangzhou 310023, Zhejiang, China
2. Shanghai Electrochemical Energy Device Research Center, Department of Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China
-
Received:2018-09-07Online:2019-07-05Published:2019-07-05 -
Contact:Guoqiang MA,Zifeng MA
摘要:
基于密度泛函理论的量子化学计算为高电压电解液的配方设计提供了理论基础。运用Gaussian软件可以有效模拟电解液中某一成分的分子构型和溶剂化状态,计算出化合物的分解路径与分解产物,进而大幅缩短电解液研发周期。本文回顾了近年来该计算方法在锂离子电池电解液研究中的相关进展,并以高电压电解液为例,介绍了该理论在溶剂氧化电位计算方面的应用,结合分子模型的优化,实现了计算值与实验值的基本统一。此外,详细阐述了砜类、氟类、离子液体等几种新型高电压溶剂和磷酸酯类、硼类、腈类等几种成膜添加剂的应用。相信今后随着动力学理论的完善和计算机技术的优化,该方法在高浓度电解液、固液界面作用机理等当前难以实现的理论模拟问题方面的应用指日可待。
中图分类号:
引用本文
李南, 马国强, 车海英, 蒋志敏, 沈旻, 董经博, 陈慧闯, 马紫峰. 密度泛函理论在高电压电解液设计中的应用[J]. 化工进展, 2019, 38(07): 3253-3264.
Nan LI, Guoqiang MA, Haiying CHE, Zhimin JIANG, Min SHEN, Jingbo DONG, Huichuang CHEN, Zifeng MA. Application of density functional theory in the design of high potential electrolyte[J]. Chemical Industry and Engineering Progress, 2019, 38(07): 3253-3264.
| 分子名称 | 分子结构 | HOMO/eV | LUMO/eV |
|---|---|---|---|
| EC |  | -8.468 | -0.604 |
| DMC |  | -8.179 | -0.370 |
| EMC |  | -8.133 | -0.256 |
| DEC |  | -8.055 | -0.268 |
表1 常见碳酸酯溶剂的HOMO和LUMO值[24]
| 分子名称 | 分子结构 | HOMO/eV | LUMO/eV |
|---|---|---|---|
| EC | | -8.468 | -0.604 |
| DMC | | -8.179 | -0.370 |
| EMC | | -8.133 | -0.256 |
| DEC | | -8.055 | -0.268 |
| 体系 | ε=1 | ε=4.2 | ε=20.5 | ε=78.4 |
|---|---|---|---|---|
| DMC/BF4 - | 4.14 | 5.79 | 6.21 | 6.29 |
| EC/BF4 - | 4.55 | 5.95 | 6.28 | 6.34 |
| PC/BF4 - | 4.57 | — | 6.25 | — |
| TMS/BF4 - | 5.23 | 6.33 | 6.49 | 6.52 |
| DMC/PF6 - | 4.56 | 6.12 | 6.51 | 6.58 |
| EC/PF6 - | 4.94 | 6.27 | 6.57 | 6.63 |
| TMS/PF6 - | 5.44 | 6.36 | 6.54 | 6.57 |
| EMS/PF6 - | 5.46 | 6.47 | 6.66 | 6.69 |
| PMS/PF6 - | 4.29 | 5.55 | 5.84 | 5.89 |
表2 不同介电常数下不同体系的氧化电位计算值单位:V[27]
| 体系 | ε=1 | ε=4.2 | ε=20.5 | ε=78.4 |
|---|---|---|---|---|
| DMC/BF4 - | 4.14 | 5.79 | 6.21 | 6.29 |
| EC/BF4 - | 4.55 | 5.95 | 6.28 | 6.34 |
| PC/BF4 - | 4.57 | — | 6.25 | — |
| TMS/BF4 - | 5.23 | 6.33 | 6.49 | 6.52 |
| DMC/PF6 - | 4.56 | 6.12 | 6.51 | 6.58 |
| EC/PF6 - | 4.94 | 6.27 | 6.57 | 6.63 |
| TMS/PF6 - | 5.44 | 6.36 | 6.54 | 6.57 |
| EMS/PF6 - | 5.46 | 6.47 | 6.66 | 6.69 |
| PMS/PF6 - | 4.29 | 5.55 | 5.84 | 5.89 |
| 分子构型 | 结合能/kJ·mol-1 | 分子构型 | 结合能/kJ·mol-1 | 分子构型 | 结合能/kJ·mol-1 |
|---|---|---|---|---|---|
| PC-Li-AsF6 | -120.5 | 2PC-Li-AsF6 | -108.9 | 3PC-Li-AsF6 | -83.4 |
| 3PC-Li-AsF6-PC | -66.7 | 3PC-Li-AsF6-2PC | -75.6 | 3PC-Li-AsF6-3PC | -94.6 |
| 3PC-Li-AsF6-4PC | -86.1 | 4PC-Li-AsF6-4PC | -117.0 | PC-Li-PF6 | -118.8 |
| 2PC-Li-PF6 | -110.3 | 3PC-Li-PF6 | -83.7 | 3PC-Li-PF6-PC | -59.3 |
| 3PC-Li-PF6-2PC | -82.7 | 3PC-Li-PF6-3PC | -92.3 | 3PC-Li-PF6-4PC | -90.7 |
| 4PC-Li-PF6-4PC | -116.5 | PC-Li-BF4 | -115.0 | 2PC-Li-BF4 | -103.0 |
| 3PC-Li-BF4 | -72.5 | 3PC-Li-BF4-PC | -64.6 | 3PC-Li-BF4-2PC | -175.9 |
| 3PC-Li-BF4-3PC | -108.5 | 3PC-Li-BF4-4PC | -89.5 | 4PC-Li-BF4-4PC | -82.4 |
| PC-Li-ClO4 | -116.8 | 2PC-Li-ClO4 | -103.5 | 3PC-Li-ClO4 | -86.5 |
| 3PC-Li-ClO4-PC | -57.9 | 3PC-Li-ClO4-2PC | -75.2 | 3PC-Li-ClO4-3PC | -100.0 |
| 3PC-Li-ClO4-4PC | -100.6 | 4PC-Li-ClO4-4PC | -97.8 |
表3 PC n -Li+-阴离子(n=1~8)的分子构型与相应结合能[28]
| 分子构型 | 结合能/kJ·mol-1 | 分子构型 | 结合能/kJ·mol-1 | 分子构型 | 结合能/kJ·mol-1 |
|---|---|---|---|---|---|
| PC-Li-AsF6 | -120.5 | 2PC-Li-AsF6 | -108.9 | 3PC-Li-AsF6 | -83.4 |
| 3PC-Li-AsF6-PC | -66.7 | 3PC-Li-AsF6-2PC | -75.6 | 3PC-Li-AsF6-3PC | -94.6 |
| 3PC-Li-AsF6-4PC | -86.1 | 4PC-Li-AsF6-4PC | -117.0 | PC-Li-PF6 | -118.8 |
| 2PC-Li-PF6 | -110.3 | 3PC-Li-PF6 | -83.7 | 3PC-Li-PF6-PC | -59.3 |
| 3PC-Li-PF6-2PC | -82.7 | 3PC-Li-PF6-3PC | -92.3 | 3PC-Li-PF6-4PC | -90.7 |
| 4PC-Li-PF6-4PC | -116.5 | PC-Li-BF4 | -115.0 | 2PC-Li-BF4 | -103.0 |
| 3PC-Li-BF4 | -72.5 | 3PC-Li-BF4-PC | -64.6 | 3PC-Li-BF4-2PC | -175.9 |
| 3PC-Li-BF4-3PC | -108.5 | 3PC-Li-BF4-4PC | -89.5 | 4PC-Li-BF4-4PC | -82.4 |
| PC-Li-ClO4 | -116.8 | 2PC-Li-ClO4 | -103.5 | 3PC-Li-ClO4 | -86.5 |
| 3PC-Li-ClO4-PC | -57.9 | 3PC-Li-ClO4-2PC | -75.2 | 3PC-Li-ClO4-3PC | -100.0 |
| 3PC-Li-ClO4-4PC | -100.6 | 4PC-Li-ClO4-4PC | -97.8 |
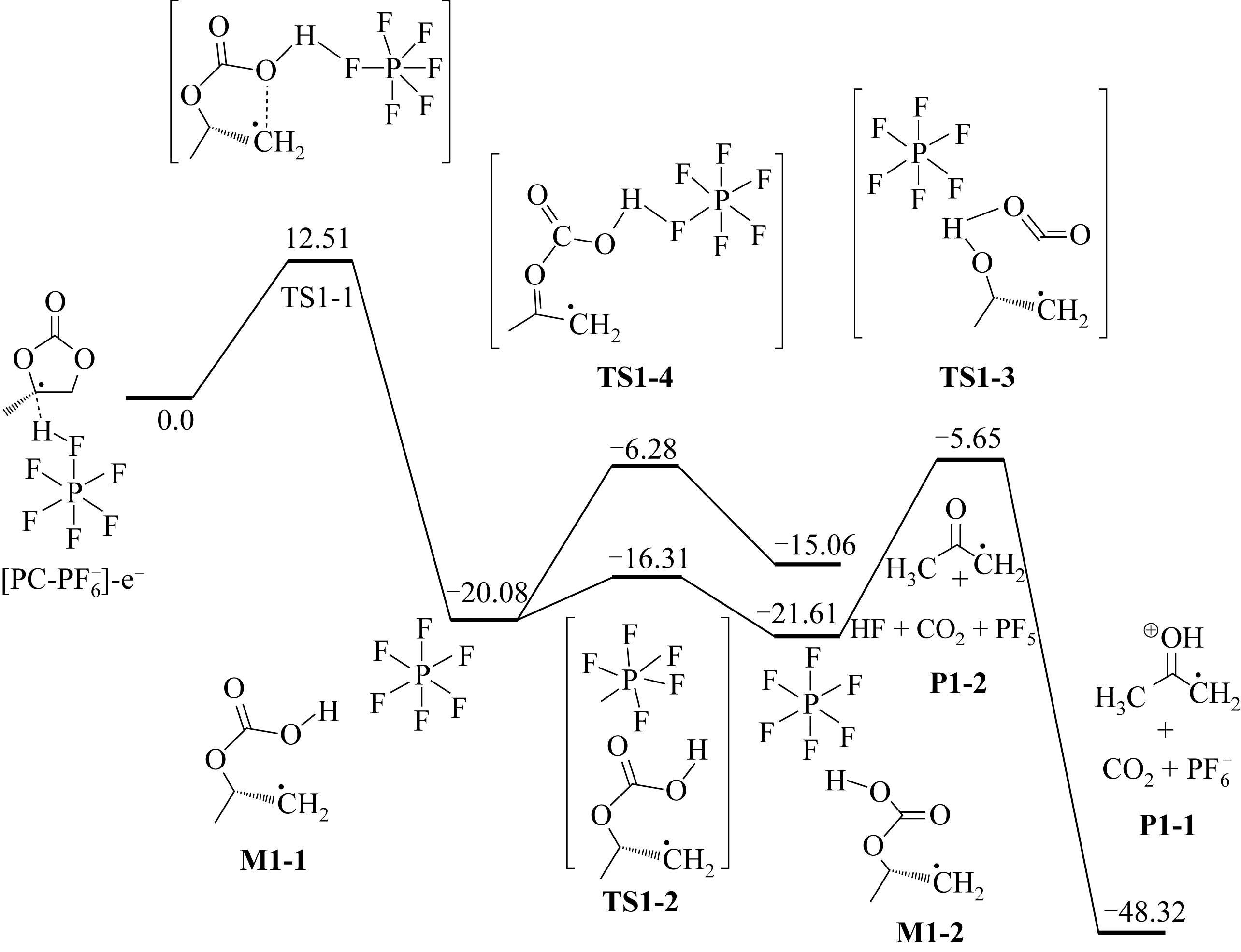
图1 PC-PF6 -的模拟氧化过程[12]

图2 碳酸酯溶剂与砜类溶剂在添加锂盐前后抗氧化能力的比较[36]

图3 正极极片表面有机分子分布示意图[37]
| 分子式 | 分子结构 | HOMO | LUMO | 理论氧化电压/V |
|---|---|---|---|---|
| EC |  | -0.31005 | -0.01067 | 6.91 |
| EMC |  | -0.29905 | 0.00251 | 6.63 |
| EPE |  | -0.26153 | 0.00596 | 5.511 |
| F-AEC | | -0.31780 | -0.01795 | 6.98 |
| F-EMC |  | -0.31946 | -0.00363 | 7.01 |
| F-EPE |  | -0.35426 | -0.00356 | 7.24 |
表4 EC、EMC和EPE氟化前后的氧化电压计算值[25]
| 分子式 | 分子结构 | HOMO | LUMO | 理论氧化电压/V |
|---|---|---|---|---|
| EC | | -0.31005 | -0.01067 | 6.91 |
| EMC | | -0.29905 | 0.00251 | 6.63 |
| EPE | | -0.26153 | 0.00596 | 5.511 |
| F-AEC | | -0.31780 | -0.01795 | 6.98 |
| F-EMC | | -0.31946 | -0.00363 | 7.01 |
| F-EPE | | -0.35426 | -0.00356 | 7.24 |
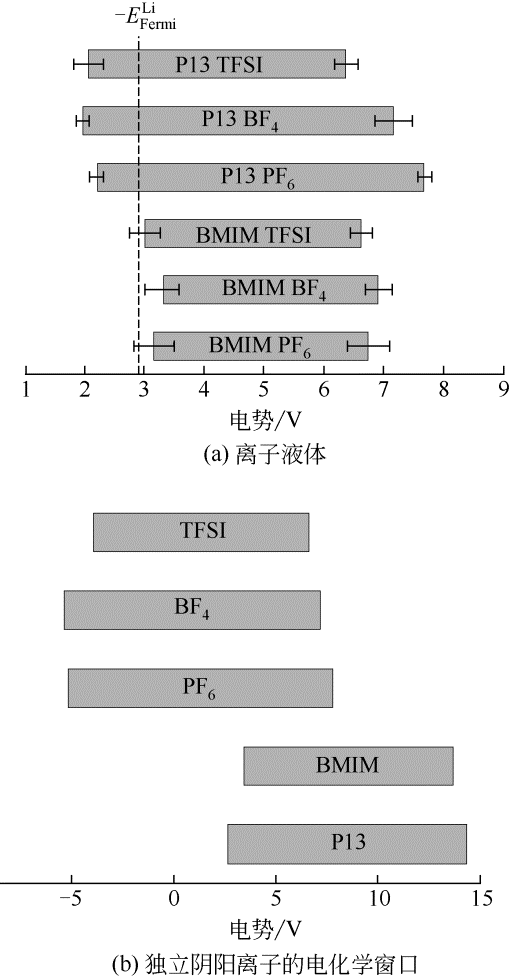
图4 离子液体和独立阴阳离子的电化学窗口计算值[43]
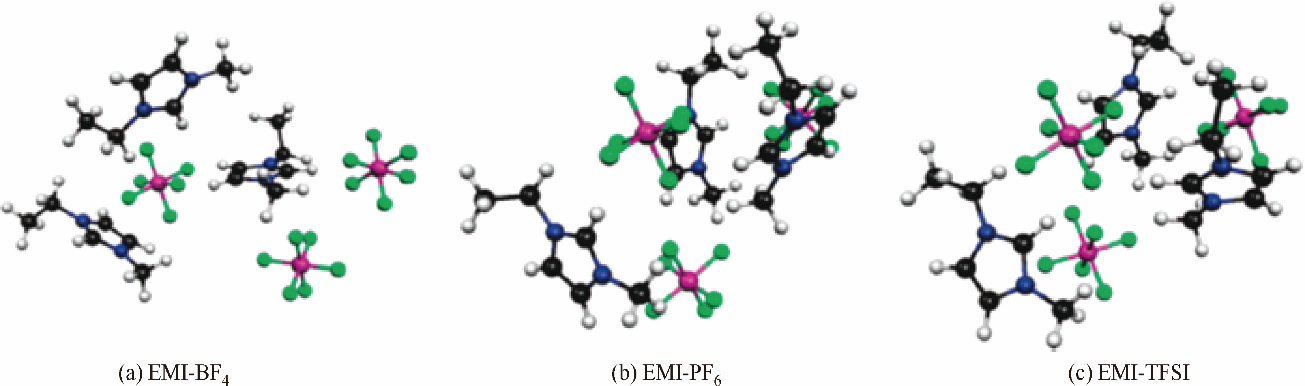
图5 EMI-BF4、EMI-PF6和EMI-TFSI最佳结构模型[44]

图6 EC、DEC、EMC、TMB、TEB和TPB的分子构型及相关氧化电位[55]

图8 EC、DMC、PTS及其相互间络合物的电解液分子氧化前后构型图与IE能量计算值 [61]
| 分子式 | 结构模型 | 氧化电位/V | PF6 -络合物氧化电位/V |
|---|---|---|---|
| TMSB |  | 5.8 | 5.7 |
| TMSP |  | 5.9 | 5.8 |
| EC |  | 7.1 | 6.5 |
| EMC |  | 6.9 | 6.5 |
表5 TMSB、TMSP、EC、EMC及其PF6 -络合物的理论氧化电位[5]
| 分子式 | 结构模型 | 氧化电位/V | PF6 -络合物氧化电位/V |
|---|---|---|---|
| TMSB | | 5.8 | 5.7 |
| TMSP | | 5.9 | 5.8 |
| EC | | 7.1 | 6.5 |
| EMC | | 6.9 | 6.5 |
| 1 | 黄学杰 . 电动汽车动力电池技术研究进展[J]. 科技导报, 2016, 34(6): 28-31. |
| HUANG X J . An overview of EVs battery technologies[J]. Science & Technology Review, 2016, 34(6): 28-31. | |
| 2 | 华政, 梁风, 姚耀春 . 电动汽车电池的发展现状与趋势[J]. 化工进展, 2017, 36(8): 2874-2881. |
| HUA Z , LIANG F , YAO Y C . Status and development trend for battery of electric vehicles[J]. Chemical Industry and Engineering Progress,2017, 36(8): 2874-2881. | |
| 3 | WANG A , KADAM S , LI H , et al . Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries[J]. npj Computational Materials, 2018, 4(15): 1-25. |
| 4 | 黄杰, 凌仕刚, 王雪龙, 等 . 锂离子电池基础科学问题——计算方法[J]. 储能科学与技术, 2015, 4(2): 215-230. |
| HUANG J , LING S G , WANG X L , et al . Fundamental scientific aspects of lithium ion batteries (ⅩⅣ)—calculation methods[J]. Energy Storage Science and Technology, 2015, 4(2): 215-230. | |
| 5 | WANG K , XING L , ZHU Y , et al . A comparative study of Si-containing electrolyte additives for lithium ion battery: which one is better and why is it better[J]. Journal of Power Sources, 2017, 342: 677-684. |
| 6 | RAKHI R , SURESH C H . A DFT study on 1,4-dihydro-1,4-azaborinine annulated linear polyacenes: absorption spectra, singlet-triplet energy gap, aromaticity, and HOMO-LUMO energy modulation[J]. Journal of Computational Chemistry, 2017, 38(26): 2232-2240. |
| 7 | CUI W , LANSAC Y , LEE H, et al . Lithium ion solvation by ethylene carbonates in lithium-ion battery electrolytes, revisited by density functional theory with the hybrid solvation model and free energy correction in solution[J]. Physical Chemistry Chemical Physics, 2016, 18: 23607-23612. |
| 8 | WANG Y , XING L , TANG X , et al . Oxidative stability and reaction mechanism of lithium bis(oxalate)borate as a cathode film-forming additive for lithium ion batteries[J]. RSC Advances, 2014, 4: 33301-33306. |
| 9 | CHEN J H , HE L M , WANG R L . Connection of DFT molecular orbital eigenvalues with the observable oxidation potentials/oxidation energies[J]. The Journal of Physical Chemistry A, 2013, 117(24): 5132-5139. |
| 10 | LIU Q , MU D , WU B , et al . Theoretical studies of the reduction of cyclic esters on the anode interface of lithium batteries[J]. Journal of the Electrochemical Society, 2017, 164(13): A3144-A3153. |
| 11 | ZHENG X , WANG X , CAI X , et al . Constructing a protective interface film on layered lithium-rich cathode using an electrolyte additive with special molecule structure[J]. ACS Applied Materials & Interfaces, 2016, 8(44): 30116-30125. |
| 12 | LEGGESSE E G , LIN R T , TENG T F , et al . Oxidative decomposition of propylene carbonate in lithium ion batteries: a DFT study[J]. The Journal of Physical Chemistry A, 2013, 117: 7959-7969. |
| 13 | CAR R, PARRINELLO M . Unified approach for molecular dynamics and density-functional theory[J]. Physical Review Letters, 1985, 55(22): 2471-2474. |
| 14 | KOZAWA T , HIROBE D , UEHARA K , et al . Low-temperature synthesis of LiNi0.5Mn1.5O4 grains using a water vaporassisted solid-state reaction[J]. Journal of Solid State Chemistry, 2018, 263: 94-99. |
| 15 | ZHOU Z F , CUI X L , ZHANG H-M , et al . Studies on Co-oxidation resistances of electrolytes based on sulfolane and lithium bis(oxalato)borate[J]. Russian Journal of Electrochemistry, 2017, 53(4): 352-358. |
| 16 | WU X , ROHMAN F , MELEDINA M , et al . Analysis of the effects of different carbon coating strategies on structure and electrochemical behavior of LiCoPO4 material as a high-voltage cathode electrode for lithium ion batteries[J]. Electrochimica Acta, 2018, 279: 108-117. |
| 17 | LIU D , ZHU W , KIM C, et al . High-energy lithium-ion battery using substituted LiCoPO4: from coin type to 1 Ah cell[J]. Journal of Power Sources, 2018, 388: 52-56. |
| 18 | LONGO R C , LIANG C , KONG F , et al . Core-shell nanocomposites for improving the structural stability of Li-rich layered oxide cathode materials for Li-ion batteries[J]. ACS Applied Materials & Interfaces, 2018, 10: 19226-19234. |
| 19 | ZUO X , ZHAO M , MA X, et al . Effect of diphenyl disulfide as an additive on the electrochemical performance of Li1.2Mn0.54Ni0.13Co0.13O2/graphite batteries at elevated temperature[J]. Electrochimica Acta, 2017, 245: 705-714. |
| 20 | ZHAO W , JI Y , ZHANG Z , et al . Recent advances in the research of functional electrolyte additives for lithium-ion batteries[J]. Current Opinion in Electrochemistry, 2017, 6(1): 84-91. |
| 21 | DELP S A , BORODIN O , OLGUIN M , et al . Importance of reduction and oxidation stability of high voltage electrolytes and additives[J]. Electrochimica Acta, 2016, 209: 498-510. |
| 22 | QIN Z , B B H, DUAN B , et al . Tributyl borate as a novel electrolyte additive to improve high voltage stability of lithium cobalt oxide in carbonate-based electrolyte[J]. Electrochimica Acta, 2018, 276: 412-416. |
| 23 | LI J , XING L , CHEN J , et al . Improving high voltage interfacial and structural stability of layered lithium-rich oxide cathode by using a boracic electrolyte additive[J]. Journal of the Electrochemical Society, 2016, 163(10): A2258-A2264. |
| 24 | WU F , ZHOU H , BAI Y , et al . Toward 5V Li-ion batteries: quantum chemical calculation and electrochemical characterization of sulfone-based high-voltage electrolytes[J]. ACS Applied Materials & Interfaces, 2015, 7(27): 15098-15107. |
| 25 | ZHANG Z C , HU L , WU H M , et al . Fluorinated electrolytes for 5V lithium-ion battery chemistry[J]. Energy & Environmental Science, 2013, 6(6): 1806-1810. |
| 26 | XING L , BORODIN O , SMITH G D , et al . Density functional theory study of the role of anions on the oxidative decomposition reaction of propylene carbonate[J]. The Journal of Physical Chemistry A, 2011, 115(47): 13896-13905. |
| 27 | BORODIN O , JOW T R . Quantum chemistry studies of the oxidative stability of carbonate, sulfone and sulfonate-based electrolytes doped with BF4 -,PF6 - anions[J]. ECS Transactions, 2011, 33(28): 77-84. |
| 28 | 邢丽丹, 杨茹, 李伟善 . 密度泛函理论方法研究锂离子电池电解液体系分子-离子结构[J]. 电化学, 2014, 20(6): 547-552. |
| XING L D , YANG R , LI W S , et al ., Density functional theory study on the structures of solvent-ion in the electrolyte of lithium ion battery[J]. Journal of Electrochemistry, 2014, 20(6): 547-552. | |
| 29 | BHATT M D , CHO M, CHO K . Interaction of Li+ ions with ethylene carbonate (EC): density functional theory calculations[J]. Applied Surface Science, 2010, 257(5): 1463-1468. |
| 30 | BORODIN O , BEHL W , JOW T R . Oxidative stability and initial decomposition reactions of carbonate, sulfone, and alkyl phosphate-based electrolytes[J]. The Journal of Physical Chemistry C, 2013, 117(17): 8661-8682. |
| 31 | GAUTHIER M , CARNEY T J , GRIMAUD A , et al . Electrode-electrolyte interface in Li-ion batteries: current understanding and new insights[J]. The Journal of Physical Chemistry Letters, 2015, 6(22): 4653-4672. |
| 32 | HAYASHI K , NEMOTO Y , TOBISHIMA S I , et al . Mixed solvent electrolyte for high voltage lithium metal secondary cells[J]. Electrochimica Acta, 1999, 44: 2337-2344. |
| 33 | XU K . Electrolytes and interphases in Li-ion batteries and beyond[J]. Chemical Reviews, 2014, 114(23): 11503-11618. |
| 34 | SHAO N , SUN X G , DAI S , et al . Electrochemical windows of sulfone-based electrolytes for high-voltage Li-ion batteries[J]. The Journal of Physical Chemistry B, 2011, 115(42): 12120-12125. |
| 35 | BHATT M D , O’DWYER C . Recent progress in theoretical and computational investigations of Li-ion battery materials and electrolytes[J]. Physical Chemistry Chemical Physics, 2015, 17: 4799-4844. |
| 36 | WANG Y , XING L , LI W , et al . Why do sulfone-based electrolytes show stability at high voltages? Insight from density functional theory[J]. The Journal of Physical Chemistry Letters, 2013, 4(22): 3992-3999. |
| 37 | XING L , VATAMANU J , BORODIN O , et al . Electrode/electrolyte interface in sulfolane-based electrolytes for Li ion batteries: a molecular dynamics simulation study[J]. The Journal of Physical Chemistry C, 2012,116(45): 23871-23881. |
| 38 | 凡俊田, 董陶, 张兰, 等 . 锂离子电池高压电解液研究进展[J]. 过程工程学报, 2018, 18(6): 1167-1177. |
| FAN J T , DONG T , ZHANG L , et al . Advances on high-voltage electrolyte of lithium ion batteries[J]. The Chinese Journal of Process Engineering, 2018, 18(6): 1167-1177. | |
| 39 | KIM C K, KIM K, SHIN K , et al . Synergistic effect of partially fluorinated ether and fluoroethylene carbonate for high-voltage lithium-ion batteries with rapid chargeability and dischargeability[J]. ACS Applied Materials & Interfaces, 2017, 9(50): 44161-44172. |
| 40 | HE M N , SU C-C , FENG Z X , et al . High voltage LiNi0.5Mn0.3Co0.2O2/graphite cell cycled at 4.6V with a FEC/HFDEC-based electrolyte[J]. Advanced Energy Materials, 2017, 7: 1700109. |
| 41 | SU C C , HE M N , REDFERN P C , et al . Oxidatively stable fluorinated sulfone electrolytes for high voltage high energy lithium-ion battery[J]. Energy & Environmental Science, 2017, 10(4): 900-904. |
| 42 | 李放放, 陈仕谋 . 高压锂离子电池电解液添加剂研究进展[J]. 储能科学与技术, 2016, 5(4): 436-442. |
| LI F F , CHEN S M . Research progress on electrolyte additives for high voltage lithium-ion batteries[J]. Energy Storage Science and Technology, 2016, 5(4): 436-442. | |
| 43 | ONG S P, ANDREUSSI O , WU Y , et al . Electrochemical windows of room-temperature ionic liquids from molecular dynamics and density functional theory calculations[J]. Chemistry of Materials, 2011, 23(11): 2979-2986. |
| 44 | ANGENENDT K , JOHANSSON P . Ionic liquid structures from large density functional theory calculations using mindless configuration[J]. The Journal of Physical Chemistry C, 2010, 114: 20577-20582. |
| 45 | ANGENENDT K , JOHANSSON P . Ionic liquid based lithium battery electrolytes: charge carriers and interactions derived by density functional theory calculations[J]. The Journal of Physical Chemistry B, 2011, 115(24): 7808-7813. |
| 46 | LOU S , QU X , MA Y, et al . Unravelling the enhanced high-temperature performance of lithium-rich oxide cathode with methyl diphenylphosphinite as electrolyte additive[J]. ChemElectroChem, 2018,5: 1569-1575. |
| 47 | HAN Y K , YOOA J , YIM T . Computational screening of phosphite derivatives as high-performance additives in high-voltage Li-ion batteries[J]. RSC Advances, 2017,7: 20049-20056. |
| 48 | LI Y , WAN S , VEITH G M , et al . A novel electrolyte salt additive for lithium-ion batteries with voltages greater than 4.7V[J]. Advanced Energy Materials, 2017,7(4): 1601397-1601403. |
| 49 | JI Y , ZHANG Z , GAO M , et al . Electrochemical behavior of suberonitrile as a high-potential electrolyte additive and Co-solvent for Li[Li0.2Mn0.56Ni0.16Co0.08]O2 cathode material[J]. Journal of the Electrochemical Society, 2015,162(4): A774-A780. |
| 50 | ZHI H , XING L , ZHENG X , et al . Understanding how nitriles stabilize electrolyte/electrode interface at high voltage[J]. The Journal of Physical Chemistry Letters, 2017, 8: 6048-6052. |
| 51 | 项宏发, 林敏, 郑浩, 等 . 环状磷酸酯作为电解液多功能添加剂的量子化学计算研究[J]. 合肥工业大学学报, 2017,40(9): 1181-1185. |
| XIANG H F , LIN M , ZHENG H , et al. Quantum chemicla calculation on cyclic phosphate compounds as multifunctional electrolyte additives[J]. Journal of Hefei University of Technology, 2017, 40(9):1181-1185. | |
| 52 | PARK M H , LEE Y S, LEE H, et al . Low Li+ binding affinity: an important characteristic for additives to form solid electrolyte interphases in Li-ion batteries[J]. Journal of Power Sources, 2011,196(11): 5109-5114. |
| 53 | LI Z D , ZHANG Y C , XIANG H F , et al . Trimethyl phosphite as an electrolyte additive for high-voltage lithium-ion batteries using lithium-rich layered oxide cathode[J]. Journal of Power Sources, 2013,240: 471-475. |
| 54 | PRAKASH REDDY V , SINN E , HOSMANE N . Boron based fluoride anion receptors: electrochemical and sensory applications[J]. Journal of Organometallic Chemistry, 2015, 798: 5-12. |
| 55 | YANG X , LI J , XING L , et al . Stabilizing lithium manganese oxide/organic carbonate electrolyte interface with a simple boron-containing additive[J], Electrochimica Acta, 2017, 227: 24-32. |
| 56 | YAMADA Y , FURUKAWA K , SODEYAMA K , et al . Unusual stability of acetonitrile-based superconcentrated electrolytes for fast-charging lithium-ion batteries[J]. Journal of the American Chemical Society, 2014, 136(13): 5039-5046. |
| 57 | AFROZ T , SEO D M, HAN S D , et al . Structural interactions within lithium salt solvates: acyclic carbonates and esters[J]. The Journal of Physical Chemistry C, 2015, 119(13): 7022-7027. |
| 58 | ZHENG X , HUANG T , PAN Y , et al . High-voltage performance of LiNi1/3Co1/3Mn1/3O2 /graphite batteries with di(methylsulfonyl) methane as a new sulfone-based electrolyte additive[J]. Journal of Power Sources, 2015, 293: 196-202. |
| 59 | WANG H , SUN D , LI X , et al . Alternative multifunctional cyclic organosilicon as an efficient electrolyte additive for high performance lithium-ion batteries[J]. Electrochimica Acta, 2017, 254: 112-122. |
| 60 | LU W , XIONG S , PU W , et al . Carbonate-grafted polysilane as a new additive for elevated-temperature lithium-ion batteries[J]. ChemElectroChem, 2017, 4(8): 2012-2018. |
| 61 | HUANG W , XING L , ZHANG R , et al . A novel electrolyte additive for improving the interfacial stability of high voltage lithium nickel manganese oxide cathode[J]. Journal of Power Sources, 2015, 293: 71-77. |
| [1] | 杨寒月, 孔令真, 陈家庆, 孙欢, 宋家恺, 王思诚, 孔标. 微气泡型下向流管式气液接触器脱碳性能[J]. 化工进展, 2023, 42(S1): 197-204. |
| [2] | 杨建平. 降低HPPO装置反应系统原料消耗的PSE[J]. 化工进展, 2023, 42(S1): 21-32. |
| [3] | 王福安. 300kt/a环氧丙烷工艺反应器降耗减排分析[J]. 化工进展, 2023, 42(S1): 213-218. |
| [4] | 王胜岩, 邓帅, 赵睿恺. 变电吸附二氧化碳捕集技术研究进展[J]. 化工进展, 2023, 42(S1): 233-245. |
| [5] | 张明焱, 刘燕, 张雪婷, 刘亚科, 李从举, 张秀玲. 非贵金属双功能催化剂在锌空气电池研究进展[J]. 化工进展, 2023, 42(S1): 276-286. |
| [6] | 时永兴, 林刚, 孙晓航, 蒋韦庚, 乔大伟, 颜彬航. 二氧化碳加氢制甲醇过程中铜基催化剂活性位点研究进展[J]. 化工进展, 2023, 42(S1): 287-298. |
| [7] | 谢璐垚, 陈崧哲, 王来军, 张平. 用于SO2去极化电解制氢的铂基催化剂[J]. 化工进展, 2023, 42(S1): 299-309. |
| [8] | 杨霞珍, 彭伊凡, 刘化章, 霍超. 熔铁催化剂活性相的调控及其费托反应性能[J]. 化工进展, 2023, 42(S1): 310-318. |
| [9] | 郑谦, 官修帅, 靳山彪, 张长明, 张小超. 铈锆固溶体Ce0.25Zr0.75O2光热协同催化CO2与甲醇合成DMC[J]. 化工进展, 2023, 42(S1): 319-327. |
| [10] | 胡喜, 王明珊, 李恩智, 黄思鸣, 陈俊臣, 郭秉淑, 于博, 马志远, 李星. 二硫化钨复合材料制备与储钠性能研究进展[J]. 化工进展, 2023, 42(S1): 344-355. |
| [11] | 戴欢涛, 曹苓玉, 游新秀, 徐浩亮, 汪涛, 项玮, 张学杨. 木质素浸渍柚子皮生物炭吸附CO2特性[J]. 化工进展, 2023, 42(S1): 356-363. |
| [12] | 张杰, 白忠波, 冯宝鑫, 彭肖林, 任伟伟, 张菁丽, 刘二勇. PEG及其复合添加剂对电解铜箔后处理的影响[J]. 化工进展, 2023, 42(S1): 374-381. |
| [13] | 高雨飞, 鲁金凤. 非均相催化臭氧氧化作用机理研究进展[J]. 化工进展, 2023, 42(S1): 430-438. |
| [14] | 顾永正, 张永生. HBr改性飞灰对Hg0的动态吸附及动力学模型[J]. 化工进展, 2023, 42(S1): 498-509. |
| [15] | 孙玉玉, 蔡鑫磊, 汤吉海, 黄晶晶, 黄益平, 刘杰. 反应精馏合成甲基丙烯酸甲酯工艺优化及节能[J]. 化工进展, 2023, 42(S1): 56-63. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||