Chemical Industry and Engineering Progress ›› 2025, Vol. 44 ›› Issue (11): 6316-6333.DOI: 10.16085/j.issn.1000-6613.2024-162
• Industrial catalysis • Previous Articles
Advances in theoretical calculation of single-atom catalysts for electrochemical nitrogen reduction to ammonia
GAN Wen( ), ZHANG Xiaofang(), JI Zhijiao, XUE Yunpeng
), ZHANG Xiaofang(), JI Zhijiao, XUE Yunpeng
- Beijing Low Carbon and Clean Energy Institute, Beijing 102200, China
-
Received:2024-10-11Revised:2025-03-27Online:2025-12-08Published:2025-11-25 -
Contact:ZHANG Xiaofang
电化学氮还原合成氨单原子催化剂的理论计算研究进展
甘汶(), 张晓方(), 纪之骄, 薛云鹏
- 北京低碳清洁能源研究院,北京 102200
-
通讯作者:张晓方 -
作者简介:甘汶(1997—),女,博士,工程师,研究方向为电化学合成氨。E-mail:20090355@ceic.com。
CLC Number:
Cite this article
GAN Wen, ZHANG Xiaofang, JI Zhijiao, XUE Yunpeng. Advances in theoretical calculation of single-atom catalysts for electrochemical nitrogen reduction to ammonia[J]. Chemical Industry and Engineering Progress, 2025, 44(11): 6316-6333.
甘汶, 张晓方, 纪之骄, 薛云鹏. 电化学氮还原合成氨单原子催化剂的理论计算研究进展[J]. 化工进展, 2025, 44(11): 6316-6333.
share this article
Add to citation manager EndNote|Ris|BibTeX
URL: https://hgjz.cip.com.cn/EN/10.16085/j.issn.1000-6613.2024-162
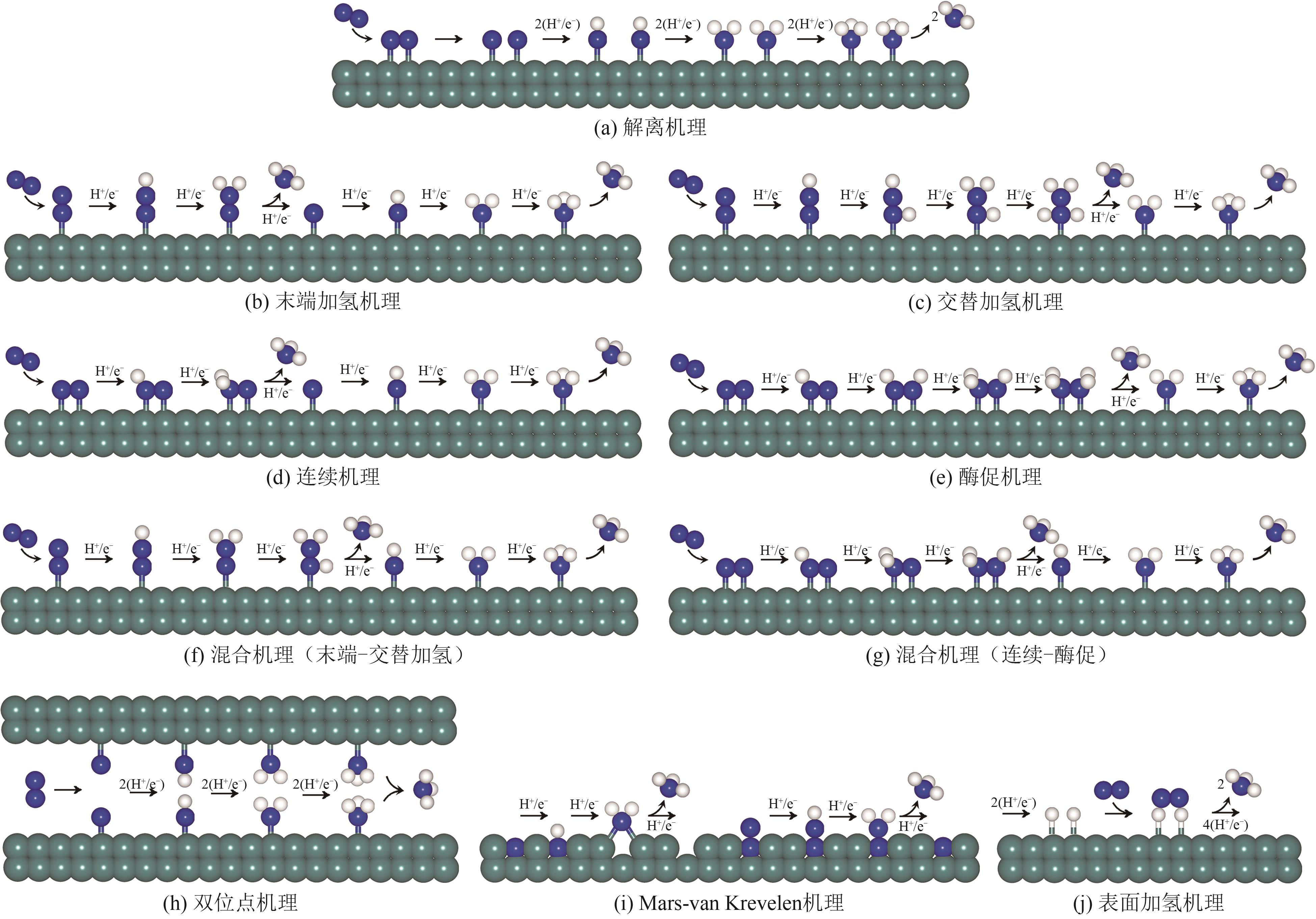
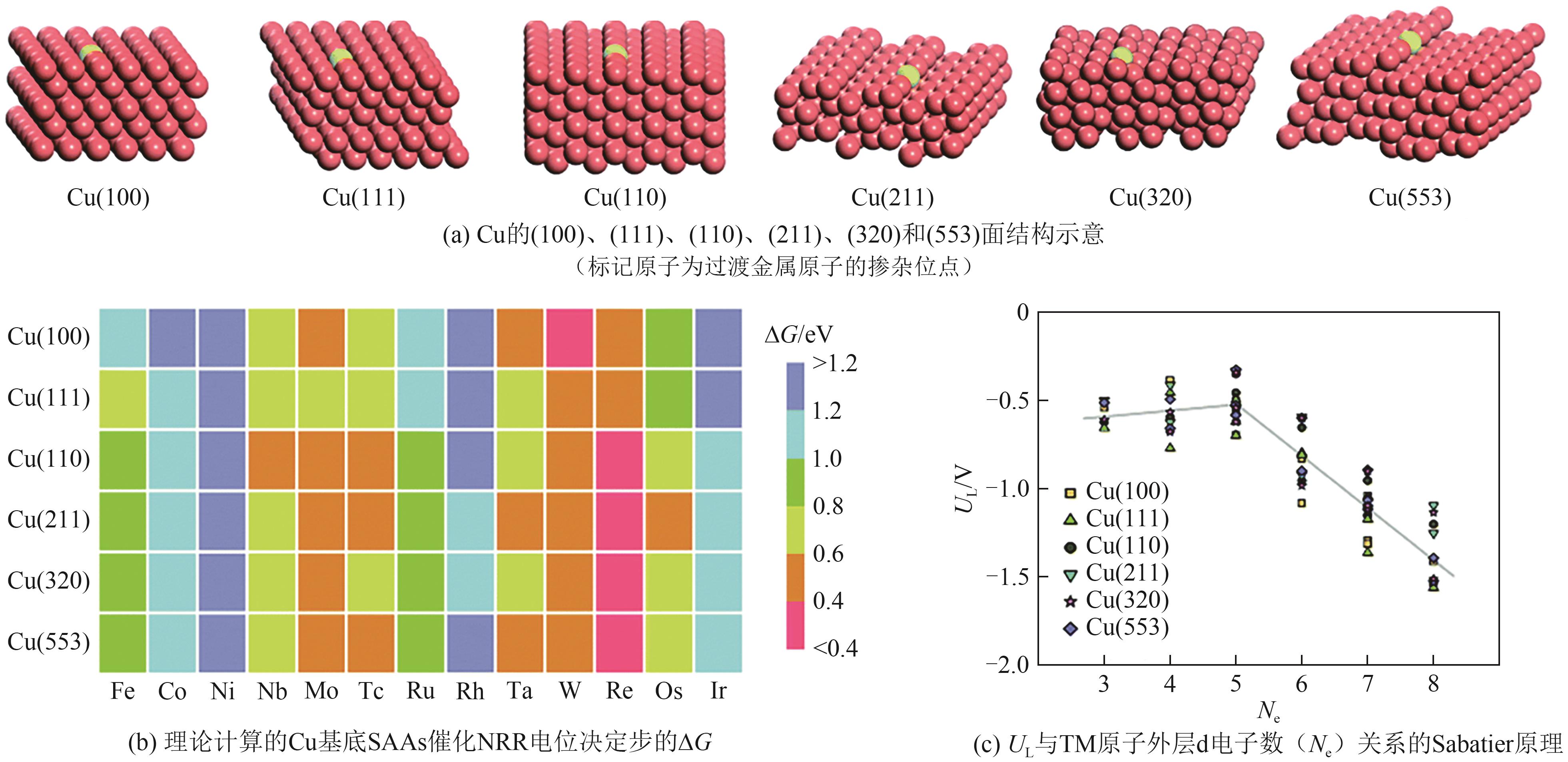
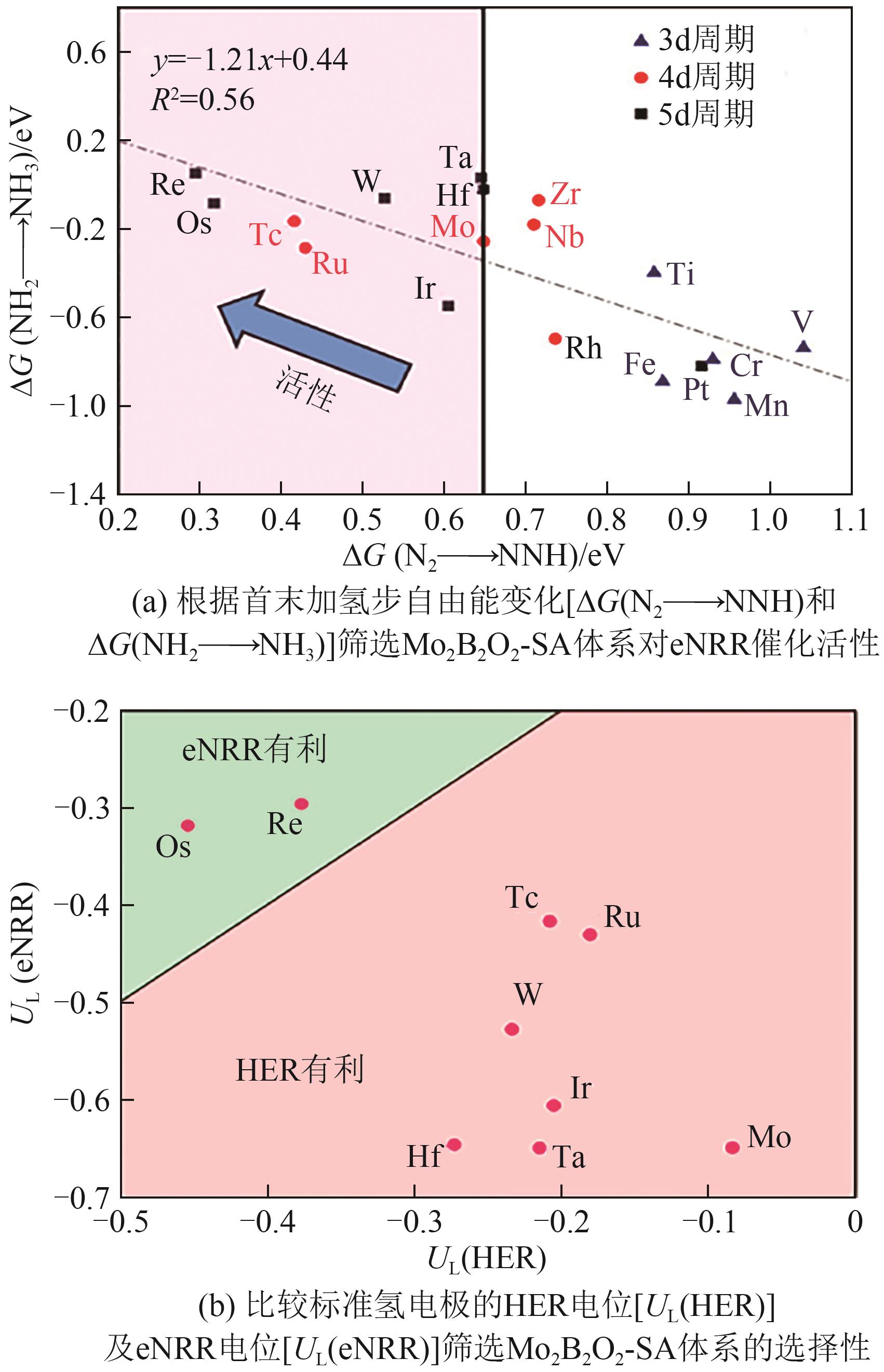
| SACs | 反应机理 | UL/V | PDS | 年份 |
|---|---|---|---|---|
| B/g-C3N4 | 酶促机理 | -0.67 | *NH2→*NH3 | 2023[ |
| Ti/CN | 酶促机理 | -0.38 | *N-*N→*N-*NH | 2021[ |
| Ti/缺陷石墨相C3N4(NVs-g-C3N4) | 酶促机理 | -0.51 | *NH2→*NH3 | 2018[ |
| V/Ti2CO2 | 连续机理 | -0.20 | *NH2→*NH3 | 2023[ |
| V/g-C3N5 | 酶促机理 | -0.30 | *N-*N→*N-*NH | 2022[ |
| V/BN | 酶促机理 | -0.41 | *NH2→*NH3 | 2019[ |
| Cr/Zr2B2O2 | 末端加氢 | -0.10 | *NH2→*NH3 | 2023[ |
| Cr/BN | 酶促机理 | -0.33 | *NH2→*NH3 | 2021[ |
| Mn/g-C9N10 | 末端加氢 | -0.30 | *N2→*NNH | 2023[ |
| Mn/Nb2CN2 | 末端加氢 | -0.51 | *N2→*NNH | 2020[ |
| Fe/过硫代苯并二氢呋喃(PTC) | 末端加氢 | -0.47 | *N2→*NNH | 2021[ |
| Fe/Nb2C | 混合机理(连续-酶促) | -0.47 | *NH2→*NH3 | 2023[ |
| Y/Mo2CO2 | 末端加氢 | -0.08 | *N2→*NNH | 2021[ |
| Y/多孔碳包覆TiO2 | 交替加氢 | -1.55 | *N2→*NNH | 2020[ |
| Zr/Mo2TiC2O2 | 末端加氢 | -0.15 | *N2→*NNH | 2019[ |
| Nb/g-C9N4 | 酶促机理 | -0.21 | *NH2→*NH3 | 2022[ |
| Nb/石墨炔 | 末端加氢 | -0.39 | *N2→*NNH | 2019[ |
| Nb/扶手椅形石墨烯带(GNR) | 混合机理(末端-交替加氢) | -0.52 | *N2→*NNH | 2022[ |
| Nb/TiO2(110) | 交替加氢 | -1.64 | *N2→*NNH | 2022[ |
| Mo/h-C4N3 | 混合机理(末端-交替加氢) | -0.18 | *N2H2→*NHNH2 | 2021[ |
| Mo/V2CO2 | 末端加氢 | -0.24 | *N2→*NNH | 2023[ |
| Mo/硫掺杂双空位石墨烯(S4-GR) | 交替加氢 | -0.29 | *N2→*NNH | 2020[ |
| Mo/57-BN | 酶促机理 | -0.29 | *N-*N→*N-*NH | 2022[ |
| Mo/Mo2CO2 | 末端加氢 | -0.32 | *N2→*NNH | 2019[ |
| Mo/Ti2NO2 | 酶促机理 | -0.32 | *N-*N→*N-*NH | 2019[ |
| Mo/单层石墨炔(GDY) | 末端加氢 | -0.33 | *N2→*NNH | 2020[ |
| Mo/B3O-石墨烯 | 酶促机理 | -0.34 | *N-*N→*N-*NH | 2021[ |
| Mo/BN | 酶促机理 | -0.35 | *NH2→*NH3 | 2017[ |
| Mo/氮掺杂石墨炔 | 末端加氢 | -0.36 | *N2→*NNH | 2022[ |
| p-Mo/四氰乙烯(TCNE) | 末端加氢 | -0.36 | *N2→*NNH | 2022[ |
| Mo/Nb2CS2 | 末端加氢 | -0.38 | *N2→*NNH | 2022[ |
| Tc/WS2 | 末端加氢 | -0.27 | *N2→*NNH | 2022[ |
| Ru/CeO2-S | 末端加氢 | -0.35 | *N2→*NNH | 2019[ |
| Ru/NC2 | 末端加氢 | -0.42 | *NNH→*NNH2 | 2019[ |
| Ru/NiO(001) | 末端加氢 | -0.49 | *NH2→*NH3 | 2023[ |
| Ta/MoS2 | 末端加氢 | -0.34 | *N2→*NNH | 2024[ |
| W/Ti2-x C2O y | 末端加氢 | -0.11 | *N2→*NNH | 2020[ |
| W/BP | 酶促机理 | -0.19 | *NH-*NH2→*NH2-*NH2 | 2021[ |
| W/MoSe2 | 末端加氢 | -0.21 | *NH2→*NH3 | 2021[ |
| W/氮掺杂多孔石墨烯 | 末端加氢 | -0.23 | *N2→*NNH | 2022[ |
| W/C9N4 | 末端加氢 | -0.25 | *N2→*NNH | 2021[ |
| W/g-CN | 末端加氢 | -0.29 | *N2→*NNH | 2020[ |
| W/g-C3N4 | 酶促机理 | -0.35 | *NH2→*NH3 | 2018[ |
| W/VSe2 | 末端加氢 | -0.36 | *N2→*NNH | 2022[ |
| Re/Mo2B2O2 | 末端加氢 | -0.29 | *N2→*NNH | 2020[ |
| Re/Cu(553) | 末端加氢 | -0.33 | *N2→*NNH | 2022[ |
| Os/B@GY | 末端加氢 | -0.32 | *NH2→*NH3 | 2023[ |
| Os@MoSe2 | 末端加氢 | -0.36 | *N2→*NNH | 2023[ |
| U/C2N | 酶促机理 | -0.44 | *N-*N→*N-*NH | 2023[ |
| SACs | 反应机理 | UL/V | PDS | 年份 |
|---|---|---|---|---|
| B/g-C3N4 | 酶促机理 | -0.67 | *NH2→*NH3 | 2023[ |
| Ti/CN | 酶促机理 | -0.38 | *N-*N→*N-*NH | 2021[ |
| Ti/缺陷石墨相C3N4(NVs-g-C3N4) | 酶促机理 | -0.51 | *NH2→*NH3 | 2018[ |
| V/Ti2CO2 | 连续机理 | -0.20 | *NH2→*NH3 | 2023[ |
| V/g-C3N5 | 酶促机理 | -0.30 | *N-*N→*N-*NH | 2022[ |
| V/BN | 酶促机理 | -0.41 | *NH2→*NH3 | 2019[ |
| Cr/Zr2B2O2 | 末端加氢 | -0.10 | *NH2→*NH3 | 2023[ |
| Cr/BN | 酶促机理 | -0.33 | *NH2→*NH3 | 2021[ |
| Mn/g-C9N10 | 末端加氢 | -0.30 | *N2→*NNH | 2023[ |
| Mn/Nb2CN2 | 末端加氢 | -0.51 | *N2→*NNH | 2020[ |
| Fe/过硫代苯并二氢呋喃(PTC) | 末端加氢 | -0.47 | *N2→*NNH | 2021[ |
| Fe/Nb2C | 混合机理(连续-酶促) | -0.47 | *NH2→*NH3 | 2023[ |
| Y/Mo2CO2 | 末端加氢 | -0.08 | *N2→*NNH | 2021[ |
| Y/多孔碳包覆TiO2 | 交替加氢 | -1.55 | *N2→*NNH | 2020[ |
| Zr/Mo2TiC2O2 | 末端加氢 | -0.15 | *N2→*NNH | 2019[ |
| Nb/g-C9N4 | 酶促机理 | -0.21 | *NH2→*NH3 | 2022[ |
| Nb/石墨炔 | 末端加氢 | -0.39 | *N2→*NNH | 2019[ |
| Nb/扶手椅形石墨烯带(GNR) | 混合机理(末端-交替加氢) | -0.52 | *N2→*NNH | 2022[ |
| Nb/TiO2(110) | 交替加氢 | -1.64 | *N2→*NNH | 2022[ |
| Mo/h-C4N3 | 混合机理(末端-交替加氢) | -0.18 | *N2H2→*NHNH2 | 2021[ |
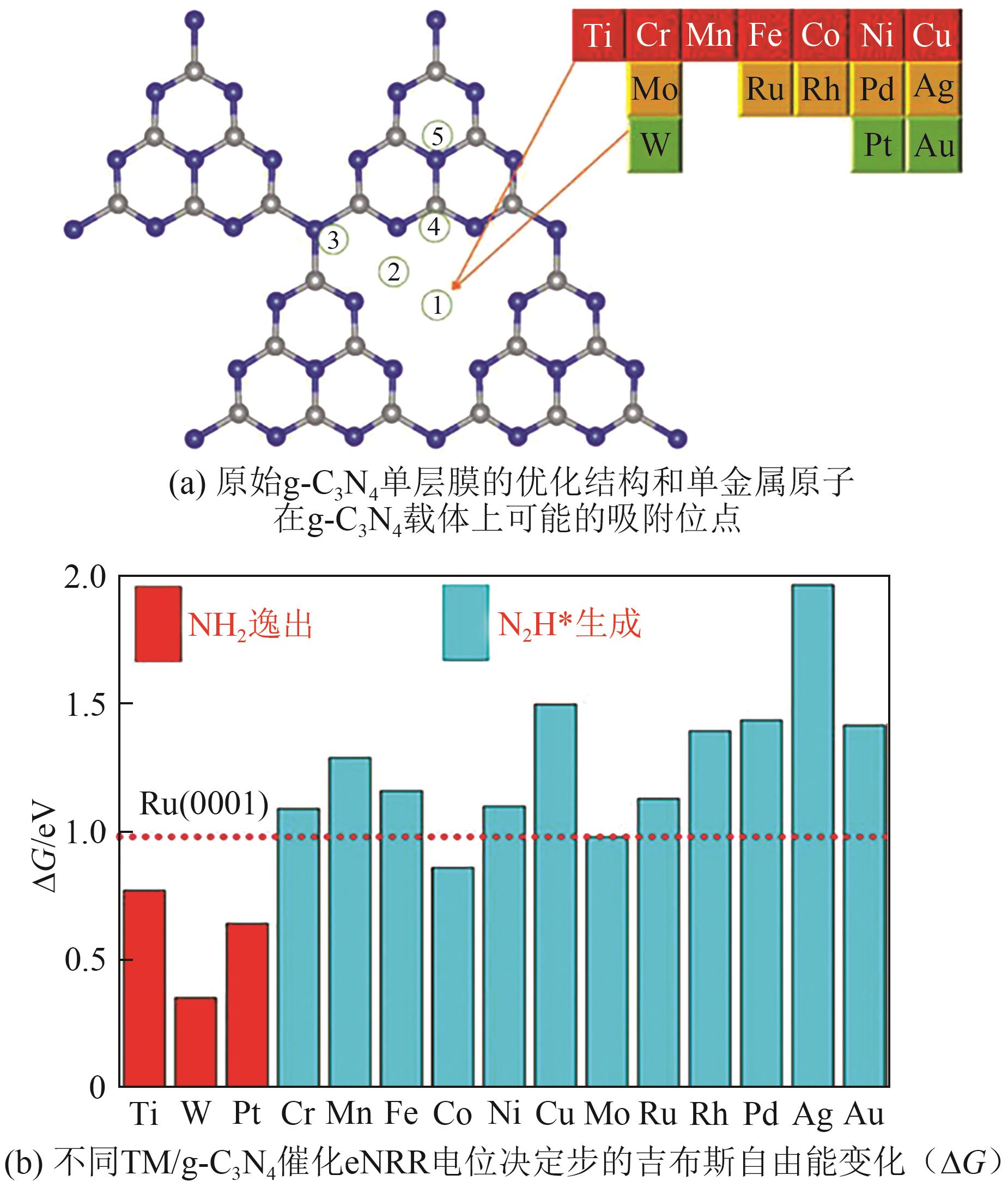
| Mo/V2CO2 | 末端加氢 | -0.24 | *N2→*NNH | 2023[ |
| Mo/硫掺杂双空位石墨烯(S4-GR) | 交替加氢 | -0.29 | *N2→*NNH | 2020[ |
| Mo/57-BN | 酶促机理 | -0.29 | *N-*N→*N-*NH | 2022[ |
| Mo/Mo2CO2 | 末端加氢 | -0.32 | *N2→*NNH | 2019[ |
| Mo/Ti2NO2 | 酶促机理 | -0.32 | *N-*N→*N-*NH | 2019[ |
| Mo/单层石墨炔(GDY) | 末端加氢 | -0.33 | *N2→*NNH | 2020[ |
| Mo/B3O-石墨烯 | 酶促机理 | -0.34 | *N-*N→*N-*NH | 2021[ |
| Mo/BN | 酶促机理 | -0.35 | *NH2→*NH3 | 2017[ |
| Mo/氮掺杂石墨炔 | 末端加氢 | -0.36 | *N2→*NNH | 2022[ |
| p-Mo/四氰乙烯(TCNE) | 末端加氢 | -0.36 | *N2→*NNH | 2022[ |
| Mo/Nb2CS2 | 末端加氢 | -0.38 | *N2→*NNH | 2022[ |
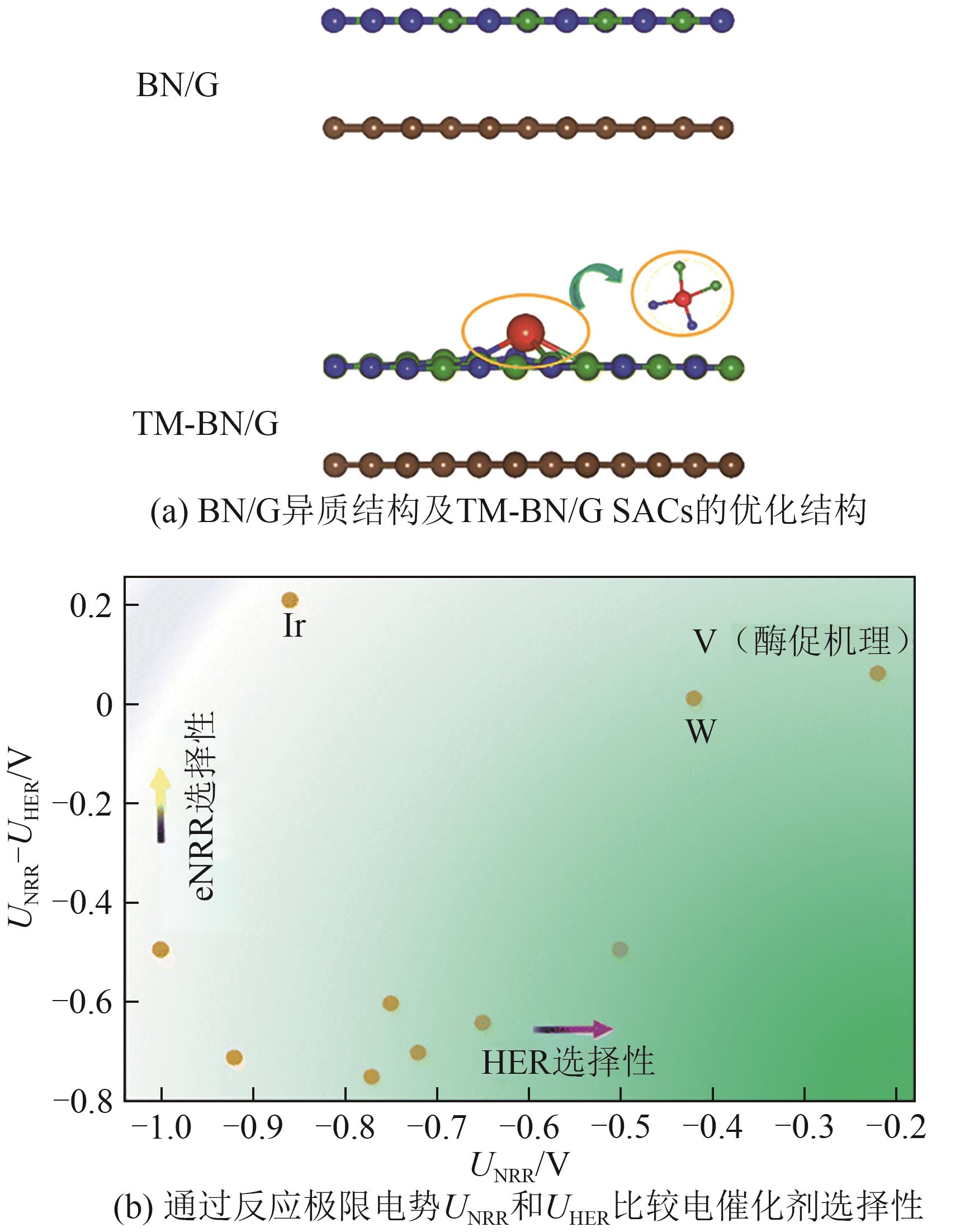
| Tc/WS2 | 末端加氢 | -0.27 | *N2→*NNH | 2022[ |
| Ru/CeO2-S | 末端加氢 | -0.35 | *N2→*NNH | 2019[ |
| Ru/NC2 | 末端加氢 | -0.42 | *NNH→*NNH2 | 2019[ |
| Ru/NiO(001) | 末端加氢 | -0.49 | *NH2→*NH3 | 2023[ |
| Ta/MoS2 | 末端加氢 | -0.34 | *N2→*NNH | 2024[ |
| W/Ti2-x C2O y | 末端加氢 | -0.11 | *N2→*NNH | 2020[ |
| W/BP | 酶促机理 | -0.19 | *NH-*NH2→*NH2-*NH2 | 2021[ |
| W/MoSe2 | 末端加氢 | -0.21 | *NH2→*NH3 | 2021[ |
| W/氮掺杂多孔石墨烯 | 末端加氢 | -0.23 | *N2→*NNH | 2022[ |
| W/C9N4 | 末端加氢 | -0.25 | *N2→*NNH | 2021[ |
| W/g-CN | 末端加氢 | -0.29 | *N2→*NNH | 2020[ |
| W/g-C3N4 | 酶促机理 | -0.35 | *NH2→*NH3 | 2018[ |
| W/VSe2 | 末端加氢 | -0.36 | *N2→*NNH | 2022[ |
| Re/Mo2B2O2 | 末端加氢 | -0.29 | *N2→*NNH | 2020[ |
| Re/Cu(553) | 末端加氢 | -0.33 | *N2→*NNH | 2022[ |
| Os/B@GY | 末端加氢 | -0.32 | *NH2→*NH3 | 2023[ |
| Os@MoSe2 | 末端加氢 | -0.36 | *N2→*NNH | 2023[ |
| U/C2N | 酶促机理 | -0.44 | *N-*N→*N-*NH | 2023[ |
| [62] | FANG Cong, AN Wei. Single-metal-atom site with high-spin state embedded in defective BN nanosheet promotes electrocatalytic nitrogen reduction[J]. Nano Research, 2021, 14(11): 4211-4219. |
| [63] | WANG Min, HUANG Yuhong, MA Fei, et al. Theoretical insights into the mechanism of nitrogen-to-ammonia electroreduction on TM/g-C9N10 [J]. Molecular Catalysis, 2023, 547: 113391. |
| [64] | KONG Youchao, LIU Di, AI Haoqiang, et al. Theoretical screening of single atoms supported on two-dimensional Nb2CN2 for nitrogen fixation[J]. ACS Applied Nano Materials, 2020, 3(11): 11274-11281. |
| [65] | LI Baoshuo, DU Wenhui, WU Qian, et al. Coronene-based 2D metal-organic frameworks: A new family of promising single-atom catalysts for nitrogen reduction reaction[J]. The Journal of Physical Chemistry C, 2021, 125(38): 20870-20876. |
| [66] | ZHANG Xuanyue, YAN Likai, SU Zhongmin. A single transition metal atom anchored on Nb2C as an electrocatalyst for the nitrogen reduction reaction[J]. Nanoscale, 2023, 15(43): 17508-17515. |
| [67] | WANG Shuo, LI Lei, HUI Kwan SAN, et al. Computational screening of single atoms anchored on defective Mo2CO2 MXene nanosheet as efficient electrocatalysts for the synthesis of ammonia[J]. Advanced Engineering Materials, 2021, 23(10): 2100405. |
| [68] | YANG Lianghao, CHOI Changhyeok, HONG Song, et al. Single yttrium sites on carbon-coated TiO2 for efficient electrocatalytic N2 reduction[J]. Chemical Communications, 2020, 56(74): 10910-10913. |
| [69] | LI Lei, WANG Xingyong, GUO Haoran, et al. Theoretical screening of single transition metal atoms embedded in MXene defects as superior electrocatalyst of nitrogen reduction reaction[J]. Small Methods, 2019, 3(11): 1900337. |
| [70] | KANG Xuxin, HUANG Junchao, DUAN Xiangmei. Computational screening of single transition-metal atoms anchored to g-C9N4 as catalysts for N2 reduction to NH3 [J]. Physical Chemistry Chemical Physics, 2022, 24(28): 17155-17162. |
| [71] | HAN Xingqi, LANG Zhongling, YAN Likai, et al. Atomic Nb anchoring on graphdiyne as a new potential electrocatalyst for nitrogen fixation: A computational view[J]. Advanced Theory and Simulations, 2019, 2(12): 1900132. |
| [72] | MA Ziyu, Peng LYU, WU Donghai, et al. V (Nb) single atoms anchored by the edge of a graphene armchair nanoribbon for efficient electrocatalytic nitrogen reduction: A theoretical study[J]. Inorganic Chemistry, 2022, 61(44): 17864-17872. |
| [73] | GAO Yunnan, YANG Yang, HAO Leiduan, et al. Single Nb atom modified anatase TiO2(110) for efficient electrocatalytic nitrogen reduction reaction[J]. Chem Catalysis, 2022, 2(9): 2275-2288. |
| [1] | HE Teng, PACHFULE Pradip, WU Hui, et al. Hydrogen carriers[J]. Nature Reviews Materials, 2016, 1(12): 16059. |
| [2] | LAN Rong, IRVINE John T S, TAO Shanwen. Ammonia and related chemicals as potential indirect hydrogen storage materials[J]. International Journal of Hydrogen Energy, 2012, 37(2): 1482-1494. |
| [3] | LIU Huazhang. Ammonia synthesis catalyst 100 years: Practice, enlightenment and challenge[J]. Chinese Journal of Catalysis, 2014, 35(10): 1619-1640. |
| [4] | ERISMAN Jan Willem, SUTTON Mark A, GALLOWAY James, et al. How a century of ammonia synthesis changed the world[J]. Nature Geoscience, 2008, 1(10): 636-639. |
| [5] | SMITH Collin, HILL Alfred K, Laura TORRENTE-MURCIANO. Current and future role of Haber-Bosch ammonia in a carbon-free energy landscape[J]. Energy & Environmental Science, 2020, 13(2): 331-344. |
| [6] | CHEN Shiming, PERATHONER Siglinda, AMPELLI Claudio, et al. Electrocatalytic synthesis of ammonia at room temperature and atmospheric pressure from water and nitrogen on a carbon-nanotube-based electrocatalyst[J]. Angewandte Chemie International Edition, 2017, 56(10): 2699-2703. |
| [7] | HUMPHREYS John, LAN Rong, TAO Shanwen. Development and recent progress on ammonia synthesis catalysts for Haber-Bosch process[J]. Advanced Energy and Sustainability Research, 2021, 2(1): 2000043. |
| [8] | 刘晓璐, 耿钰晓, 郝然, 等. 环境条件下电催化氮还原的现状、挑战与展望[J]. 化学进展, 2021, 33(7): 1074-1091. |
| LIU Xiaolu, GENG Yuxiao, HAO Ran, et al. Electrocatalytic nitrogen reduction reaction under ambient condition: Current status, challenges, and perspectives[J]. Progress in Chemistry, 2021, 33(7): 1074-1091. | |
| [9] | ZHANG Shuai, ZHAO Yunxuan, SHI Run, et al. Photocatalytic ammonia synthesis: Recent progress and future[J]. EnergyChem, 2019, 1(2): 100013. |
| [10] | RAO Li, XU Xin, ADAMO Carlo. Theoretical investigation on the role of the central carbon atom and close protein environment on the nitrogen reduction in Mo nitrogenase[J]. ACS Catalysis, 2016, 6(3): 1567-1577. |
| [11] | SHIPMAN Michael A, SYMES Mark D. Recent progress towards the electrosynthesis of ammonia from sustainable resources[J]. Catalysis Today, 2017, 286: 57-68. |
| [74] | AGARWAL Sakshi, KUMAR Ritesh, ARYA Rakesh, et al. Rational design of single-atom catalysts for enhanced electrocatalytic nitrogen reduction reaction[J]. The Journal of Physical Chemistry C, 2021, 125(23): 12585-12593. |
| [75] | FU Zhanzhao, WU Mingliang, LI Qiang, et al. A simple descriptor for the nitrogen reduction reaction over single atom catalysts[J]. Materials Horizons, 2023, 10(3): 852-858. |
| [76] | QIN Yanyang, WU Deyin, SU Yaqiong. The effect of grain boundary in hexagonal boron nitride on catalytic activity of nitrogen reduction reaction[J]. Applied Surface Science, 2022, 593: 153468. |
| [77] | HUANG Bin, LI Neng, Wee-Jun ONG, et al. Single atom-supported MXene: How single-atomic-site catalysts tune the high activity and selectivity of electrochemical nitrogen fixation[J]. Journal of Materials Chemistry A, 2019, 7(48): 27620-27631. |
| [78] | CHENG Yuwen, DAI Jianhong, SONG Yan, et al. Single molybdenum atom anchored on 2D Ti2NO2 MXene as a promising electrocatalyst for N2 fixation[J]. Nanoscale, 2019, 11(39): 18132-18141. |
| [79] | ZHAI Xingwu, YAN Hongxia, GE Guixian, et al. The single-Mo-atom-embedded-graphdiyne monolayer with ultra-low onset potential as high efficient electrocatalyst for N2 reduction reaction[J]. Applied Surface Science, 2020, 506: 144941. |
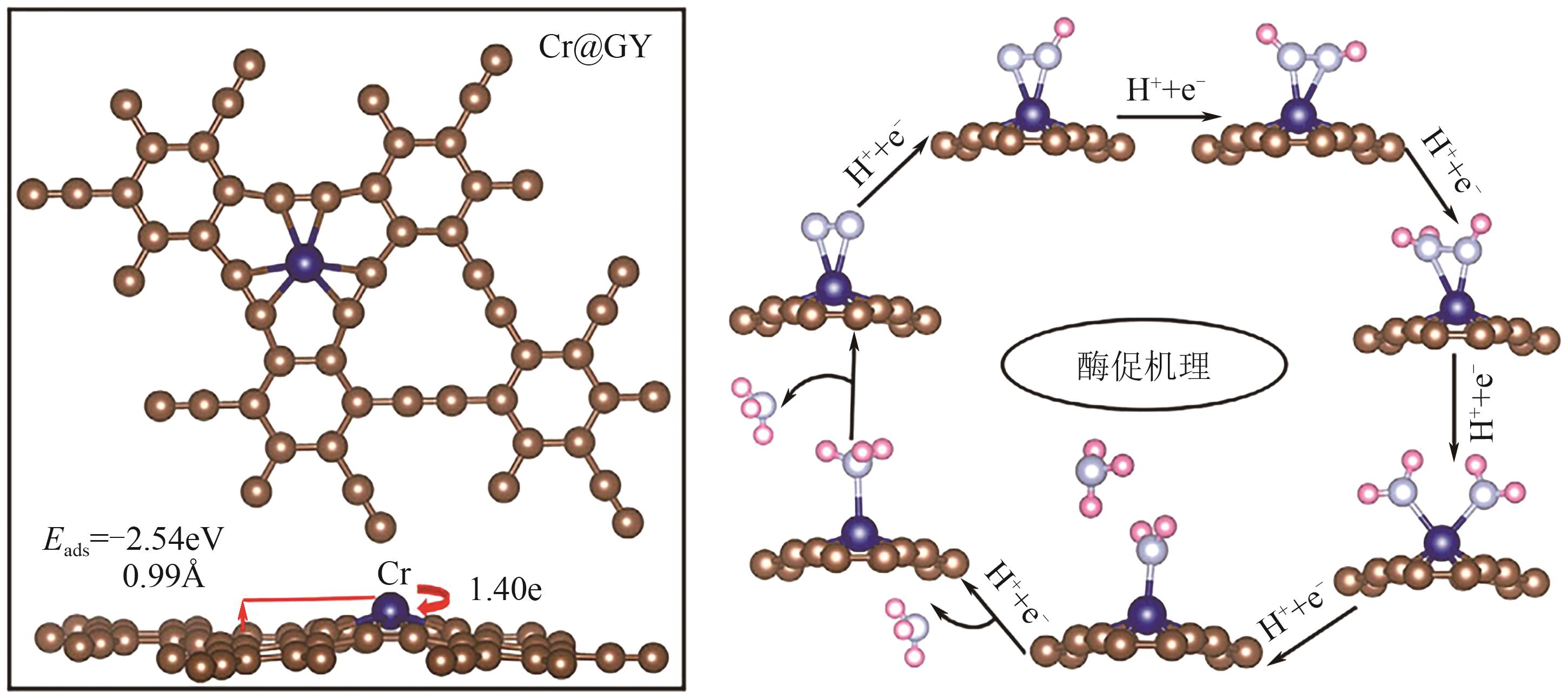
| [80] | KONG Lingyi, QIU Siyao, CAI Qinghai, et al. Boosting nitrogen reduction on single Mo atom by tuning its coordination environment[J]. Sustainable Energy & Fuels, 2021, 5(24): 6488-6497. |
| [81] | RU Sen, HE Mingqi, ZHOU Yanan, et al. Theoretical and comparative analysis of graphdiyne and confined flexible nitrogen-doped graphdiyne-supported single-atom catalysts for electrochemical nitrogen reduction[J]. The Journal of Physical Chemistry C, 2022, 126(43): 18282-18291. |
| [82] | DENG Dan, SONG Bingyi, LI Cong, et al. High-throughput screening of transition-metal-atom-embedded parallel tetracyanoethylene 2D networks as single-atom electrocatalysts for ammonia synthesis and its underlying microscopic mechanisms[J]. The Journal of Physical Chemistry C, 2022, 126(49): 20816-20830. |
| [83] | HUANG Bin, YANG Jing, REN Guangyuan, et al. Design of single-atom catalysts on S-functionalized Mxenes for enhanced activity and selectivity in N2 electroreduction[J]. Applied Catalysis A: General, 2022, 646: 118886. |
| [84] | CHEN Yibo, ZHANG Xinyu, QIN Jiaqian, et al. Taming the challenges of activity and selectivity in catalysts for electrochemical N2 fixation via single metal atom supported on WS2 [J]. Applied Surface Science, 2022, 571: 151357. |
| [85] | QI Jiamin, GAO Liye, WEI Fenfei, et al. Design of a high-performance electrocatalyst for N2 conversion to NH3 by trapping single metal atoms on stepped CeO2 [J]. ACS Applied Materials & Interfaces, 2019, 11(50): 47525-47534. |
| [12] | CAO Na, ZHENG Gengfeng. Aqueous electrocatalytic N2 reduction under ambient conditions[J]. Nano Research, 2018, 11(6): 2992-3008. |
| [13] | 詹溯, 章福祥. 常温常压电催化合成氨的研究进展[J]. 化学学报, 2021, 79(2): 146-157. |
| ZHAN Su, ZHANG Fuxiang. Recent progress on electrocatalytic synthesis of ammonia under amibent conditions[J]. Acta Chimica Sinica, 2021, 79(2): 146-157. | |
| [14] | TAYYEBI Ebrahim, ABGHOUI Younes, Egill SKÚLASON. Elucidating the mechanism of electrochemical N2 reduction at the Ru(0001) electrode[J]. ACS Catalysis, 2019, 9(12): 11137-11145. |
| [15] | LING Chongyi, ZHANG Yehui, LI Qiang, et al. New mechanism for N2 reduction: The essential role of surface hydrogenation[J]. Journal of the American Chemical Society, 2019, 141(45): 18264-18270. |
| [16] | ISHIKAWA Atsushi, Toshiki DOI, NAKAI Hiromi. Catalytic performance of Ru, Os, and Rh nanoparticles for ammonia synthesis: A density functional theory analysis[J]. Journal of Catalysis, 2018, 357: 213-222. |
| [17] | MONTOYA Joseph H, TSAI Charlie, VOJVODIC Aleksandra, et al. The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations[J]. ChemSusChem, 2015, 8(13): 2180-2186. |
| [18] | GARDEN Anna L, Egill SKÚLASON. The mechanism of industrial ammonia synthesis revisited: Calculations of the role of the associative mechanism[J]. The Journal of Physical Chemistry C, 2015, 119(47): 26554-26559. |
| [19] | HELLMAN A, BAERENDS E J, BICZYSKO M, et al. Predicting catalysis: Understanding ammonia synthesis from first-principles calculations[J]. The Journal of Physical Chemistry B, 2006, 110(36): 17719-17735. |
| [20] | LOGADÓTTIR Á, NØRSKOV J K. Ammonia synthesis over a Ru(0001) surface studied by density functional calculations[J]. Journal of Catalysis, 2003, 220(2): 273-279. |
| [21] | ROD T H, LOGADOTTIR A, NØRSKOV J K. Ammonia synthesis at low temperatures[J]. The Journal of Chemical Physics, 2000, 112(12): 5343-5347. |
| [22] | CHEN Z W, LANG X Y, JIANG Q. Discovery of cobweb-like MoC6 and its application for nitrogen fixation[J]. Journal of Materials Chemistry A, 2018, 6(20): 9623-9628. |
| [23] | ZHONG Yuan, ZHAO Xiaojie, FENG Yuliang, et al. DFT study on the electrochemical synthesis of ammonia over Mo2C(121) with N-doping[J]. Molecular Catalysis, 2022, 530: 112637. |
| [24] | ELLINGSSON Viktor, IQBAL Atef, Egill SKÚLASON, et al. Nitrogen reduction reaction to ammonia on transition metal carbide catalysts[J]. ChemSusChem, 2023, 16(22): e202300947. |
| [86] | TAO Hengcong, CHOI Changhyeok, DING Liangxin, et al. Nitrogen fixation by Ru single-atom electrocatalytic reduction[J]. Chem, 2019, 5(1): 204-214. |
| [87] | HUANG Xiang, WANG Jiong, ZHAO Changming, et al. NiO matrix decorated by Ru single atoms: Electron-rich Ru-induced high activity and selectivity toward electrochemical N2 reduction[J]. The Journal of Physical Chemistry Letters, 2023, 14(16): 3785-3793. |
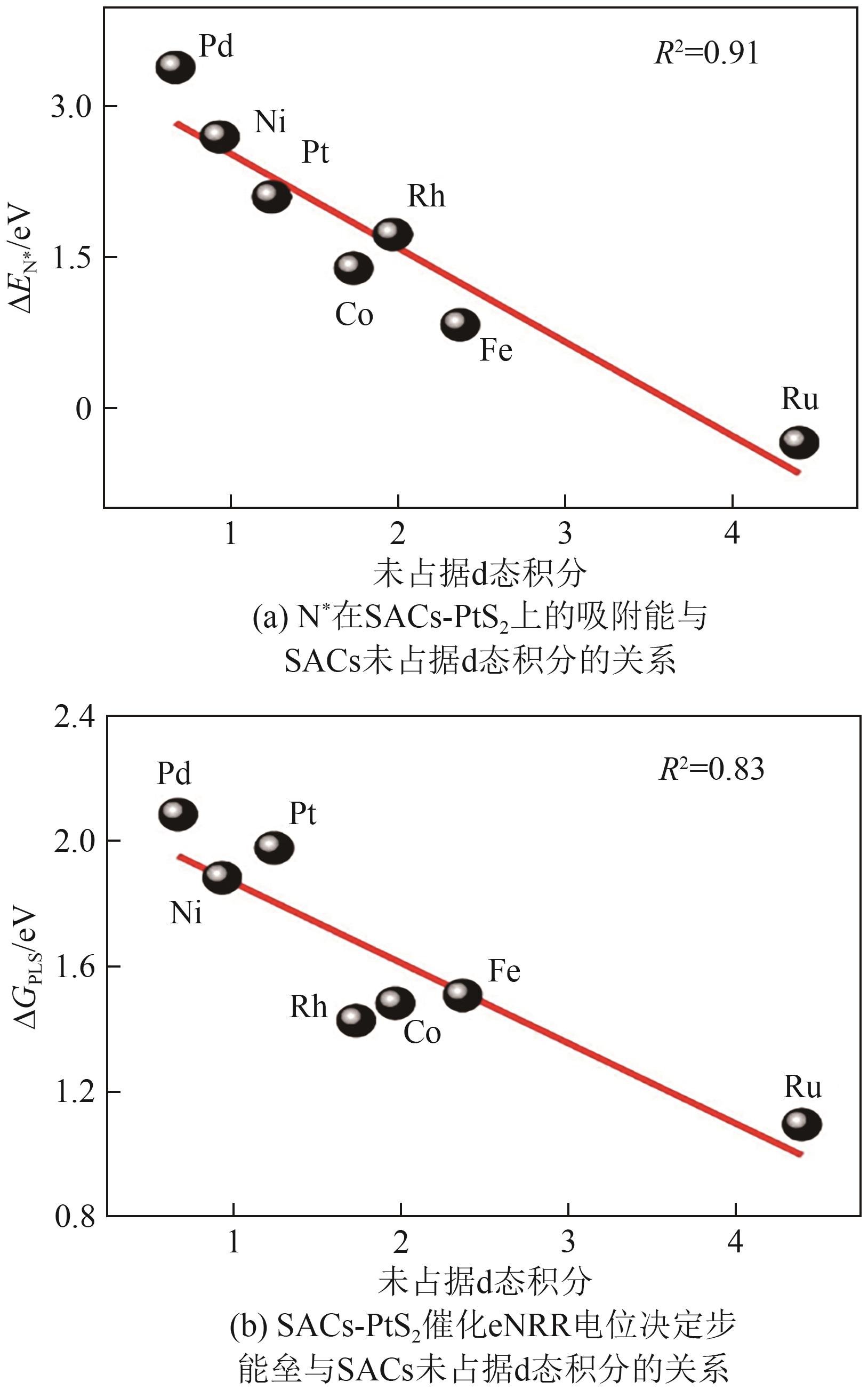
| [88] | BAI Zhiqiang, LIU Yufang, ZHANG Wenhua. Synergistic multi-adsorption of N2 on MoS2 supported single metal atoms: Enhancing electroreduction of nitrogen to ammonia[J]. Journal of Physics and Chemistry of Solids, 2024, 184: 111664. |
| [89] | TANG Shaobin, LIU Tianyong, DANG Qian, et al. Synergistic effect of surface-terminated oxygen vacancy and single-atom catalysts on defective MXenes for efficient nitrogen fixation[J]. The Journal of Physical Chemistry Letters, 2020, 11(13): 5051-5058. |
| [90] | WU Jie, LI Jiahui, YU Yangxin. Single Nb or W atom-embedded BP monolayers as highly selective and stable electrocatalysts for nitrogen fixation with low-onset potentials[J]. ACS Applied Materials & Interfaces, 2021, 13(8): 10026-10036. |
| [91] | CHEN Can, CAO Juexian, YIN Wenjin, et al. Single transition metal atom modified MoSe2 as a promising electrocatalyst for nitrogen fixation: A first-principles study[J]. Chemical Physics Letters, 2021, 780: 138939. |
| [92] | LIU Shize, LIU Jingyao. Rational design of highly efficient electrocatalytic single-atom catalysts for nitrogen reduction on nitrogen-doped graphene and g-C2N supports[J]. Journal of Power Sources, 2022, 535: 231449. |
| [93] | MENG Qingling, ZHANG Ling, WU Jinge, et al. First-principles screening of single transition metal atoms anchored on two-dimensional C9N4 for the nitrogen reduction reaction[J]. Physical Chemistry Chemical Physics, 2021, 23(14): 8784-8791. |
| [94] | NIU Huan, WANG Xiting, SHAO Chen, et al. Computational screening single-atom catalysts supported on g-CN for N2 reduction: High activity and selectivity[J]. ACS Sustainable Chemistry & Engineering, 2020, 8(36): 13749-13758. |
| [95] | CHEN Zhe, ZHAO Jingxiang, CABRERA Carlos R, et al. Computational screening of efficient single-atom catalysts based on graphitic carbon nitride (g-C3N4) for nitrogen electroreduction[J]. Small Methods, 2019, 3(6): 1800368. |
| [96] | WANG Jiahui, LUO Zhifen, ZHANG Xicheng, et al. Single transition metal atom anchored on VSe2 as electrocatalyst for nitrogen reduction reaction[J]. Applied Surface Science, 2022, 580: 152272. |
| [97] | YAO Mengkai, SHI Zuhao, ZHANG Peng, et al. Density functional theory study of single metal atoms embedded into MBene for electrocatalytic conversion of N2 to NH3 [J]. ACS Applied Nano Materials, 2020, 3(10): 9870-9879. |
| [98] | DAI Tianyi, WANG Zhili, LANG Xingyou, et al. “Sabatier principle” of d electron number for describing the nitrogen reduction reaction performance of single-atom alloy catalysts[J]. Journal of Materials Chemistry A, 2022, 10(32): 16900-16907. |
| [99] | SONG Wei, FU Zhe, MA Pengfei, et al. Theoretical insights into nonmetal-doped graphyne-supported noble metal electrocatalysts for NH3 synthesis via nitrogen reduction[J]. Applied Surface Science, 2023, 617: 156550. |
| [100] | HOU Pengfei, HUANG Yuhong, MA Fei, et al. Screening of single-atom catalysts of transition metal supported on MoSe2 for high-efficiency nitrogen reduction reaction[J]. Molecular Catalysis, 2023, 537: 112967. |
| [101] | LIU Huijie, QU Mengnan, DU Aijun, et al. N2 reduction in uranium-doped C2N/C3N4 monolayers: A DFT computational study[J]. New Journal of Chemistry, 2023, 47(29): 13880-13887. |
| [102] | BECKE A D, EDGECOMBE K E. A simple measure of electron localization in atomic and molecular systems[J]. The Journal of Chemical Physics, 1990, 92(9): 5397-5403. |
| [103] | DERINGER Volker L, TCHOUGRÉEFF Andrei L, DRONSKOWSKI Richard. Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets[J]. The Journal of Physical Chemistry A, 2011, 115(21): 5461-5466. |
| [104] | HENKELMAN Graeme, ARNALDSSON Andri, Hannes JÓNSSON. A fast and robust algorithm for Bader decomposition of charge density[J]. Computational Materials Science, 2006, 36(3): 354-360. |
| [105] | HU Xiuli, XIONG Lixin, FANG Weihai, et al. Computational insight into metallated graphynes as single atom electrocatalysts for nitrogen fixation[J]. ACS Applied Materials & Interfaces, 2022, 14(24): 27861-27872. |
| [106] | TANG Shaobin, DANG Qian, LIU Tianyong, et al. Realizing a not-strong-not-weak polarization electric field in single-atom catalysts sandwiched by boron nitride and graphene sheets for efficient nitrogen fixation[J]. Journal of the American Chemical Society, 2020, 142(45): 19308-19315. |
| [107] | ZHANG Hongchao, CUI Chaonan, LUO Zhixun. MoS2-supported Fe2 clusters catalyzing nitrogen reduction reaction to produce ammonia[J]. The Journal of Physical Chemistry C, 2020, 124(11): 6260-6266. |
| [108] | ZHANG Lifu, ZHAO Wanghui, ZHANG Wenhua, et al. gt-C3N4 Coordinated single atom as an efficient electrocatalyst for nitrogen reduction reaction[J]. Nano Research, 2019, 12(5): 1181-1186. |
| [109] | BO Tingting, CAO Shiqian, MU Nan, et al. High-throughput screening of transition metal single-atom catalysts for nitrogen reduction reaction[J]. Nano Energy 2020, 68: 104304. |
| [25] | LI Qinye, HE Lizhong, SUN Chenghua, et al. Computational study of MoN2 monolayer as electrochemical catalysts for nitrogen reduction[J]. The Journal of Physical Chemistry C, 2017, 121(49): 27563-27568. |
| [26] | KONG Youchao, HE Tianwei, PUENTE SANTIAGO Alain R, et al. Unravelling the reaction mechanisms of N2 fixation on molybdenum nitride: A full DFT study from the pristine surface to heteroatom anchoring[J]. ChemSusChem, 2021, 14(16): 3257-3266. |
| [27] | HÖSKULDSSON Árni B, ABGHOUI Younes, GUNNARSDÓTTIR Anna B, et al. Computational screening of rutile oxides for electrochemical ammonia formation[J]. ACS Sustainable Chemistry & Engineering, 2017, 5(11): 10327-10333. |
| [28] | WANG Jiaqi, JIANG Zhou, PENG Guiming, et al. Surface valence state effect of MoO2+ x on electrochemical nitrogen reduction[J]. Advanced Science, 2022, 9(12): 2104857. |
| [29] | LEE Chi Ho, PAHARI Silabrata, SITAPURE Niranjan, et al. Investigating high-performance non-precious transition metal oxide catalysts for nitrogen reduction reaction: A multifaceted DFT-kMC-LSTM approach[J]. ACS Catalysis, 2023, 13(13): 8336-8346. |
| [30] | YAO Xue, CHEN Zhiwen, WANG Yaru, et al. Activated basal planes of WS2 by intrinsic defects as catalysts for the electrocatalytic nitrogen reduction reaction[J]. Journal of Materials Chemistry A, 2019, 7(45): 25961-25968. |
| [31] | WEN Lu, REN Chunjin, ZOU Yu, et al. Why it is S-rich around Mo atom in the nitrogenase: A DFT investigation[J]. Applied Surface Science, 2020, 534: 147595. |
| [32] | GIUFFREDI Giorgio, ASSET Tristan, LIU Yuanchao, et al. Transition metal chalcogenides as a versatile and tunable platform for catalytic CO2 and N2 electroreduction[J]. ACS Materials Au, 2021, 1(1): 6-36. |
| [33] | MAIBAM Ashakiran, BABARAO Ravichandar, KRISHNAMURTY Sailaja. Doped 2D VX2 (X = S, Se, Te) monolayers as electrocatalysts for ammonia production: A DFT based study[J]. Applied Surface Science, 2022, 602: 154401. |
| [34] | Thi H HO, Viet Q BUI, NGUYEN Quynh Anh T, et al. Unleashing the power of boron: Enhancing nitrogen reduction reaction through defective ReS2 monolayers[J]. Physical Chemistry Chemical Physics, 2023, 25(37): 25389-25397. |
| [35] | ZHAO Yan, NIU Xueqing, GUO Fengjuan, et al. First-principles study on the performance of electrocatalytic nitrogen reduction reaction on MoB2(001) surface[J]. Journal of Physics and Chemistry of Solids, 2023, 181: 111483. |
| [36] | GHOSHAL Sourav, GHOSH Atish, ROY Prodyut, et al. Recent progress in computational design of single-atom/cluster catalysts for electrochemical and solar-driven N2 fixation[J]. ACS Catalysis, 2022, 12(24): 15541-15575. |
| [37] | IQBAL Muhammad Saqlain, YAO Zhibo, RUAN Yukun, et al. Single-atom catalysts for electrochemical N2 reduction to NH3 [J]. Rare Metals, 2023, 42(4): 1075-1097. |
| [110] | XU Ziwei, SONG Ruofei, WANG Mingyuan, et al. Single atom-doped arsenene as electrocatalyst for reducing nitrogen to ammonia: A DFT study[J]. Physical Chemistry Chemical Physics, 2020, 22(45): 26223-26230. |
| [111] | MEDFORD Andrew J, VOJVODIC Aleksandra, HUMMELSHØJ Jens S, et al. From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis[J]. Journal of Catalysis, 2015, 328: 36-42. |
| [112] | GAO Shuaishuai, MA Zuju, XIAO Chengwei, et al. High-throughput computational screening of single-atom embedded in defective BN nanotube for electrocatalytic nitrogen fixation[J]. Applied Surface Science, 2022, 591: 153130. |
| [113] | GE Lei, XU Weiwei, CHEN Chongyang, et al. Rational prediction of single metal atom supported on two-dimensional metal diborides for electrocatalytic N2 reduction reaction with integrated descriptor[J]. The Journal of Physical Chemistry Letters, 2020, 11(13): 5241-5247. |
| [114] | LIAO Xiaobin, LU Ruihu, XIA Lixue, et al. Density functional theory for electrocatalysis[J]. Energy & Environmental Materials, 2022, 5(1): 157-185. |
| [115] | CHEN Siyu, GAO Yongqi, WANG Wugang, et al. Prediction of three-metal cluster catalysts on two-dimensional W2N3 support with integrated descriptors for electrocatalytic nitrogen reduction[J]. ACS Nano, 2023, 17(2): 1522-1532. |
| [116] | WANG Shuyue, QIAN Chao, ZHOU Shaodong. Accelerating the development of electrocatalysts for electrochemical nitrogen fixation through theoretical and computational approaches[J]. Materials Chemistry Frontiers, 2023, 7(19): 4259-4280. |
| [117] | SHU Zheng, YAN Hejin, CHEN Hongfei, et al. Mutual modulation via charge transfer and unpaired electrons of catalytic sites for the superior intrinsic activity of N2 reduction: From high-throughput computation assisted with a machine learning perspective[J]. Journal of Materials Chemistry A, 2022, 10(10): 5470-5478. |
| [118] | SUN Jingchao, ZHENG Dian, DENG Fei, et al. Heterogeneous N-heterocyclic carbenes supported single-atom catalysts for nitrogen fixation: A combined density functional theory and machine learning study[J]. Applied Surface Science, 2024, 644: 158802. |
| [119] | YANG Ze, GAO Wang. Applications of machine learning in alloy catalysts: Rational selection and future development of descriptors[J]. Advanced Science, 2022, 9(12): 2106043. |
| [120] | CHEN Zhiwen, LU Zhuole, CHEN Lixin, et al. Machine-learning-accelerated discovery of single-atom catalysts based on bidirectional activation mechanism[J]. Chem Catalysis, 2021, 1(1): 183-195. |
| [121] | ZAFARI Mohammad, NISSIMAGOUDAR Arun S, UMER Muhammad, et al. First principles and machine learning based superior catalytic activities and selectivities for N2 reduction in MBenes, defective 2D materials and 2D π-conjugated polymer-supported single atom catalysts[J]. Journal of Materials Chemistry A, 2021, 9(14): 9203-9213. |
| [38] | JING Wentong, SHEN Hui, QIN Ruixuan, et al. Surface and interface coordination chemistry learned from model heterogeneous metal nanocatalysts: From atomically dispersed catalysts to atomically precise clusters[J]. Chemical Reviews, 2023, 123(9): 5948-6002. |
| [39] | YANG Di, LI Jinsheng, XIAO Meiling, et al. atomically dispersed metal catalysts towards nitrogen reduction for ammonia: From homogeneous to heterogeneous[J]. Chemical Engineering Journal, 2023, 468: 143776. |
| [40] | 纪之骄, 张晓方, 甘汶, 等. 载体对单原子电催化剂合成氨性能的影响与调控策略[J]. 化工学报, 2025, 76(1): 18-39. |
| JI Zhijiao, ZHANG Xiaofang, GAN Wen, et al. Influence of support on the performance of single atom electrocatalyst for ammonia synthesis and the control strategy[J]. CIESC Journal, 2025, 76(1): 18-39. | |
| [41] | LONG Xianhu, HUANG Fan, YAO Zhangnan, et al. Advancements in electrocatalytic nitrogen reduction: A comprehensive review of single-atom catalysts for sustainable ammonia synthesis[J]. Small, 2024, 20(32): 2400551. |
| [42] | GUO Haoran, ZHANG Haotian, ZHAO Jiayang, et al. Two-dimensional WO3-transition-metal dichalcogenide vertical heterostructures for nitrogen fixation: A photo(electro) catalysis theoretical strategy[J]. The Journal of Physical Chemistry C, 2022, 126(6): 3043-3053. |
| [43] | JI Shuang, WANG Zhongxu, ZHAO Jingxiang. A boron-interstitial doped C2N layer as a metal-free electrocatalyst for N2 fixation: A computational study[J]. Journal of Materials Chemistry A, 2019, 7(5): 2392-2399. |
| [44] | MAO Xin, ZHOU Si, YAN Cheng, et al. A single boron atom doped boron nitride edge as a metal-free catalyst for N2 fixation[J]. Physical Chemistry Chemical Physics, 2019, 21(3): 1110-1116. |
| [45] | CAO Yongyong, DENG Shengwei, FANG Qiaojun, et al. Single and double boron atoms doped nanoporous C2N-h2D electrocatalysts for highly efficient N2 reduction reaction: A density functional theory study[J]. Nanotechnology, 2019, 30(33): 335403. |
| [46] | CHEN Z W, CHEN L X, YANG C C, et al. Atomic (single, double, and triple atoms) catalysis: Frontiers, opportunities, and challenges[J]. Journal of Materials Chemistry A, 2019, 7(8): 3492-3515. |
| [47] | WANG Yongxia, CUI Xiangzhi, ZHANG Jinqiang, et al. Advances of atomically dispersed catalysts from single-atom to clusters in energy storage and conversion applications[J]. Progress in Materials Science, 2022, 128: 100964. |
| [48] | YANG Xuan, KATTEL Shyam, NASH Jared, et al. Quantification of active sites and elucidation of the reaction mechanism of the electrochemical nitrogen reduction reaction on vanadium nitride[J]. Angewandte Chemie International Edition, 2019, 58(39): 13768-13772. |
| [49] | ABGHOUI Younes, GARDEN Anna L, HOWALT Jakob G, et al. Electroreduction of N2 to ammonia at ambient conditions on mononitrides of Zr, Nb, Cr, and V: A DFT guide for experiments[J]. ACS Catalysis, 2016, 6(2): 635-646. |
| [50] | ZHAO Jingxiang, CHEN Zhongfang. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study[J]. Journal of the American Chemical Society, 2017, 139(36): 12480-12487. |
| [122] | WANG Shuyue, QIAN Chao, ZHOU Shaodong. Machine learning design of single-atom catalysts for nitrogen fixation[J]. ACS Applied Materials & Interfaces, 2023, 15(34): 40656-40664. |
| [123] | ZHANG Lifu, CHEN Lanlan, ZHAO Wanghui, et al. Self-promoted ammonia selectivity for the electro-reduction of nitrogen on gt-C3N4 supported single metal catalysts: The machine learning model and physical insights[J]. Inorganic Chemistry Frontiers, 2023, 10(22): 6578-6587. |
| [124] | QIAO Botao, WANG Aiqin, YANG Xiaofeng, et al. Single-atom catalysis of CO oxidation using Pt1/FeO x [J]. Nature Chemistry, 2011, 3(8): 634-641. |
| [125] | Robert SCHLÖGL. Catalytic synthesis of ammonia—A “never-ending story”?[J]. Angewandte Chemie International Edition, 2003, 42(18): 2004-2008. |
| [126] | WANG Ziqiang, LI Yinghao, YU Hongjie, et al. Ambient electrochemical synthesis of ammonia from nitrogen and water catalyzed by flower-like gold microstructures[J]. ChemSusChem, 2018, 11(19): 3480-3485. |
| [127] | CHEN Qianqian, ZHANG Xiaodong, JIN Yuwei, et al. An overview on noble metal (group Ⅷ)-based heterogeneous electrocatalysts for nitrogen reduction reaction[J]. Chemistry—An Asian Journal, 2020, 15(24): 4131-4152. |
| [128] | KYRIAKOU Georgios, BOUCHER Matthew B, JEWELL April D, et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations[J]. Science, 2012, 335(6073): 1209-1212. |
| [129] | ZHANG Tianjun, WALSH Andrew G, YU Jihong, et al. Single-atom alloy catalysts: Structural analysis, electronic properties and catalytic activities[J]. Chemical Society Reviews, 2021, 50(1): 569-588. |
| [130] | GREINER M T, JONES T E, BEEG S, et al. Free-atom-like d states in single-atom alloy catalysts[J]. Nature Chemistry, 2018, 10(10): 1008-1015. |
| [131] | CAO Ning, ZHANG Nan, QIU Yongqing, et al. Electroreduction of N2 to NH3 catalyzed by a Mn/Re(111) single-atom alloy catalyst with high activity and selectivity: A new insight from a first-principles study[J]. Catalysis Science & Technology, 2022, 12(12): 4074-4085. |
| [132] | LIU P, NØRSKOV J K. Ligand and ensemble effects in adsorption on alloy surfaces[J]. Physical Chemistry Chemical Physics, 2001, 3(17): 3814-3818. |
| [133] | SONG Wei, WANG Mengmeng, LIU Xiao, et al. Mo-embedded Ir-based electrocatalyst for nitrogen reduction reaction: A computational study[J]. Materials Today Communications, 2022, 33: 104318. |
| [51] | ZHANG Ning, WANG Mei, LIU Jingya. Two-dimensional conductive covalent organic framework for efficient electrocatalytic nitrogen reduction reaction[J]. Vacuum, 2023, 210: 111852. |
| [52] | WANG Tao, Frank ABILD-PEDERSEN. Achieving industrial ammonia synthesis rates at near-ambient conditions through modified scaling relations on a confined dual site[J]. Proceedings of the National Academy of Sciences of the United States of America, 2021, 118(30): e2106527118. |
| [53] | ABGHOUI Younes, Egill SKÚLASSON. Transition metal nitride catalysts for electrochemical reduction of nitrogen to ammonia at ambient conditions[J]. Procedia Computer Science, 2015, 51: 1897-1906. |
| [54] | PAN Jaysree, HANSEN Heine Anton, VEGGE Tejs. Vanadium oxynitrides as stable catalysts for electrochemical reduction of nitrogen to ammonia: The role of oxygen[J]. Journal of Materials Chemistry A, 2020, 8(45): 24098-24107. |
| [55] | RASOOL Anjumun, ANIS Insha, AHMAD BHAT Sajad, et al. Optimizing the NRR activity of single and double boron atom catalysts using a suitable support: A first principles investigation[J]. Physical Chemistry Chemical Physics, 2023, 25(33): 22275-22285. |
| [56] | CHU Zhaoqin, KANG Xuxin, DUAN Xiangmei. Single metal atom anchored on a CN monolayer as an excellent electrocatalyst for the nitrogen reduction reaction[J]. Physical Chemistry Chemical Physics, 2021, 23(4): 2658-2662. |
| [57] | CHEN Xingzhu, ZHAO Xiujian, KONG Zhouzhou, et al. Unravelling the electrochemical mechanisms for nitrogen fixation on single transition metal atoms embedded in defective graphitic carbon nitride[J]. Journal of Materials Chemistry A, 2018, 6(44): 21941-21948. |
| [58] | HU Jinnian, TIAN Lingchan, WANG Haiyan, et al. Theoretical screening of single-atom electrocatalysts of MXene-supported 3d-metals for efficient nitrogen reduction[J]. Chinese Journal of Catalysis, 2023, 52: 252-262. |
| [59] | QUAN Chuye, XIAO Shanshan, YI Yingwei, et al. Explore the underlying mechanism of graphitic C3N5-hosted single-atom catalyst for electrocatalytic nitrogen fixation[J]. International Journal of Hydrogen Energy, 2022, 47(52): 22035-22044. |
| [60] | MA Zuju, CUI Zhitao, XIAO Chengwei, et al. Theoretical screening of efficient single-atom catalysts for nitrogen fixation based on a defective BN monolayer[J]. Nanoscale, 2020, 12(3): 1541-1550. |
| [61] | GAO Ya, WANG Erpeng, ZHENG Yazhuo, et al. Hexagonal MBenes-supported single atom as electrocatalysts for the nitrogen reduction reaction[J]. Energy Material Advances, 2023, 4: 39. |
| [134] | ZHANG Linlin, CONG Meiyu, DING Xin, et al. A Janus Fe-SnO2 catalyst that enables bifunctional electrochemical nitrogen fixation[J]. Angewandte Chemie International Edition, 2020, 59(27): 10888-10893. |
| [135] | YAO Zhibo, LIU Shiqiang, LIU Honghong, et al. Pre-adsorbed H-assisted N2 activation on single-atom cadmium-O5 decorated In2O3 for efficient NH3 electrosynthesis[J]. Advanced Functional Materials, 2023, 33(5): 2209843. |
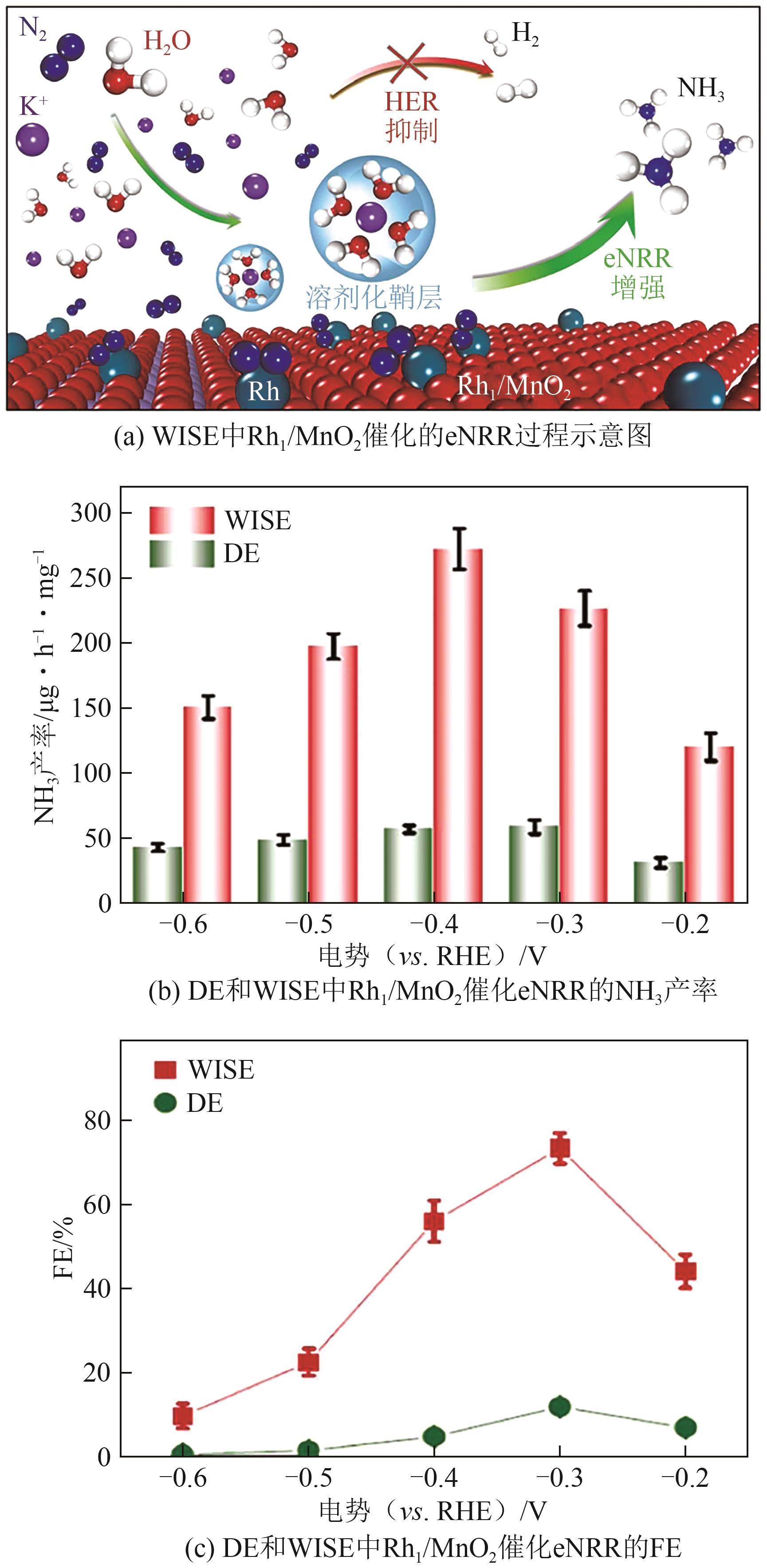
| [136] | SHEN Peng, LI Xiaotian, LUO Yaojing, et al. Ultra-efficient N2 electroreduction achieved over a rhodium single-atom catalyst (Rh1/MnO2) in water-in-salt electrolyte[J]. Applied Catalysis B: Environmental, 2022, 316: 121651. |
| [137] | WANKE Sieghard E, FLYNN Peter C. The sintering of supported metal catalysts[J]. Catalysis Reviews, 1975, 12(1): 93-135. |
| [138] | O’CONNOR Nolan J, JONAYAT A S M, JANIK Michael J, et al. Interaction trends between single metal atoms and oxide supports identified with density functional theory and statistical learning[J]. Nature Catalysis, 2018, 1: 531-539. |
| [139] | ZHAO Xunhua, LEVELL Zachary H, YU Saerom, et al. Atomistic understanding of two-dimensional electrocatalysts from first principles[J]. Chemical Reviews, 2022, 122(12): 10675-10709. |
| [140] | NOVOSELOV K S, GEIM A K, MOROZOV S V, et al. Electric field effect in atomically thin carbon films[J]. Science, 2004, 306(5696): 666-669. |
| [141] | CHITTIBABU Dinesh Kumar Dhanthala, SATHISHKUMAR Nadaraj, WU Shiuan-Yau, et al. Single-atom metal anchored penta-graphene for highly efficient and selective electroreduction of nitrogen into ammonia[J]. ACS Applied Energy Materials, 2023, 6(12): 6636-6645. |
| [142] | CHOI Changhyeok, BACK Seoin, KIM Na-Young, et al. Suppression of hydrogen evolution reaction in electrochemical N2 reduction using single-atom catalysts: A computational guideline[J]. ACS Catalysis, 2018, 8(8): 7517-7525. |
| [143] | ZHAO Wanghui, ZHANG Lifu, LUO Qiquan, et al. Single Mo1(Cr1) atom on nitrogen-doped graphene enables highly selective electroreduction of nitrogen into ammonia[J]. ACS Catalysis, 2019, 9(4): 3419-3425. |
| [144] | QU Mengnan, QIN Gangqiang, FAN Jianfen, et al. Theoretical insights into the performance of single and double transition metal atoms doped on N-graphenes for N2 electroreduction[J]. Applied Surface Science, 2021, 537: 148012. |
| [145] | SENTHAMARAIKANNAN Thillai Govindaraja, KALIAPERUMAL Selvaraj, KRISHNAMURTY Sailaja. Role of chemical structure of support in enhancing the catalytic activity of a single atom catalyst toward NRR: A computational study[J]. Frontiers in Chemistry, 2021, 9: 733422. |
| [146] | CHEN Shaona, BU Mengke, ZHOU Zhangyu, et al. Boosting nitrogen reduction to ammonia on Fe-N3S sites by introduction S into defect graphene[J]. Materials Today Energy, 2022, 25: 100954. |
| [147] | ZHANG Yaqin, WANG Yuhang, MA Ninggui, et al. Establishing an orbital-level understanding of active origins of heteroatom-coordinated single-atom catalysts: The case of N2 reduction[J]. Journal of Colloid and Interface Science, 2023, 650: 961-971. |
| [148] | GAO Zhengyang, HUANG Hanyu, XU Shaopeng, et al. Regulating the coordination environment through doping N atoms for single-atom Mn electrocatalyst of N2 reduction with high catalytic activity and selectivity: A theoretical study[J]. Molecular Catalysis, 2020, 493: 111091. |
| [149] | QIN Yanyang, ZHANG Shishi, GAO Guoxin, et al. A theoretical study on molybdenum and sulfur Co-doped graphene for electrocatalytic nitrogen reduction[J]. Molecular Catalysis, 2022, 517: 112048. |
| [150] | SONG Wei, XIE Kun, GUO Yongliang, et al. Computational screening of 3d transition metal atoms anchored on defective graphene for efficient electrocatalytic N2 fixation[J]. ChemPhysChem, 2021, 22(16): 1712-1721. |
| [151] | DENG Dehui, NOVOSELOV K S, FU Qiang, et al. Catalysis with two-dimensional materials and their heterostructures[J]. Nature Nanotechnology, 2016, 11(3): 218-230. |
| [152] | TANG Yanan, CHEN Weiguang, ZHAO Mingyu, et al. Theoretical prediction on stability, electronic and activity properties of single-atom catalysts anchored graphene and boron phosphide heterostructures[J]. Fuel, 2023, 332: 126213. |
| [153] | LI Deqing, GAO Jinai, HAO Mingtian, et al. Electrochemical mechanisms of the reduction of N2 to NH3 catalyzed by TM anchored on Nvs-g-C3N4/graphene van der Waals heterostructure: Insights from a first-principles study[J]. Molecular Catalysis, 2023, 547: 113353. |
| [154] | SONG Wei, FU Ling, HE Chaozheng, et al. Carbon-coordinated single Cr site for efficient electrocatalytic N2 fixation[J]. Advanced Theory and Simulations, 2021, 4(6): 2100044. |
| [155] | HE Junjie, MA Shuangying, ZHOU Pan, et al. Magnetic properties of single transition-metal atom absorbed graphdiyne and graphyne sheet from DFT+U calculations[J]. The Journal of Physical Chemistry C, 2012, 116(50): 26313-26321. |
| [156] | YU Huidi, XUE Yurui, HUANG Bolong, et al. Ultrathin nanosheet of graphdiyne-supported palladium atom catalyst for efficient hydrogen production[J]. iScience, 2019, 11: 31-41. |
| [157] | FENG Zhen, TANG Yanan, CHEN Weiguang, et al. Graphdiyne coordinated transition metals as single-atom catalysts for nitrogen fixation[J]. Physical Chemistry Chemical Physics, 2020, 22(17): 9216-9224. |
| [158] | TAN Li, NIE Chunyang, AO Zhimin, et al. Novel two-dimensional crystalline carbon nitrides beyond g-C3N4: Structure and applications[J]. Journal of Materials Chemistry A, 2021, 9(1): 17-33. |
| [159] | CHEN Yibo, ZHANG Xinyu, QIN Jiaqian, et al. Theoretical screening of highly efficient single-atom catalysts for nitrogen reduction based on a defective C3N monolayer[J]. International Journal of Hydrogen Energy, 2022, 47(8): 5292-5306. |
| [160] | LI Xin, ZHOU Qianyu, WANG Shifeng, et al. Tuning the coordination environment to effect the electrocatalytic behavior of a single-atom catalyst toward the nitrogen reduction reaction[J]. The Journal of Physical Chemistry C, 2021, 125(22): 11963-11974. |
| [161] | DUTTA Supriti, PATI Swapan K. Novel design of single transition metal atoms anchored on C6N6 nanosheet for electrochemical and photochemical N2 reduction to ammonia[J]. Catalysis Today, 2023, 424: 113804. |
| [162] | REN Chunjin, JIANG Qianyu, LIN Wei, et al. Density functional theory study of single-atom V, Nb, and Ta catalysts on graphene and carbon nitride for selective nitrogen reduction[J]. ACS Applied Nano Materials, 2020, 3(6): 5149-5159. |
| [163] | QIAN Yumin, LIU Yuanyue, ZHAO Yu, et al. Single vs double atom catalyst for N2 activation in nitrogen reduction reaction: A DFT perspective[J]. EcoMat, 2020, 2(1): e12014. |
| [164] | LI Hao, YU Bing, ZHUANG Zechao, et al. A small change in the local atomic environment for a big improvement in single-atom catalysis[J]. Journal of Materials Chemistry A, 2021, 9(7): 4184-4192. |
| [165] | LIU Anmin, LIANG Xingyou, REN Xuefeng, et al. Recent progress in MXene-based materials: Potential high-performance electrocatalysts[J]. Advanced Functional Materials, 2020, 30(38): 2003437. |
| [166] | ANASORI Babak, SHI Chenyang, MOON Eun Ju, et al. Control of electronic properties of 2D carbides (MXenes) by manipulating their transition metal layers[J]. Nanoscale Horizons, 2016, 1(3): 227-234. |
| [167] | BU Fanxing, ZAGHO Moustafa M, IBRAHIM Yasseen, et al. Porous MXenes: Synthesis, structures, and applications[J]. Nano Today, 2020, 30: 100803. |
| [168] | 景远聚, 康淳, 林延欣, 等. MXene基单原子催化剂的制备及其在电催化中的应用[J]. 化学进展, 2022, 34(11): 2373-2385. |
| JING Juyuan, KANG Chun, LIN Yanxin, et al. MXene-based single-atom catalysts: Synthesis and electrochemical catalysis[J]. Progress in Chemistry, 2022, 34(11): 2373-2385. | |
| [169] | NIU Kaifeng, CHI Lifeng, ROSEN Johanna, et al. Termination-accelerated electrochemical nitrogen fixation on single-atom catalysts supported by MXenes[J]. The Journal of Physical Chemistry Letters, 2022, 13(12): 2800-2807. |
| [170] | LUO Heng, WANG Xiaoxu, WAN Chubin, et al. A theoretical study of Fe adsorbed on pure and nonmetal (N, F, P, S, Cl)-doped Ti3C2O2 for electrocatalytic nitrogen reduction[J]. Nanomaterials, 2022, 12(7): 1081. |
| [171] | XU Tianjie, WANG Yuhua, XIONG Zuzhao, et al. A rising 2D star: Novel MBenes with excellent performance in energy conversion and storage[J]. Nano-Micro Letters, 2022, 15(1): 6. |
| [172] | HAO Yu, XU Lichun, PU Jibin, et al. Stable zigzag edges of transition-metal dichalcogenides with high catalytic activity for oxygen reduction[J]. Electrochimica Acta, 2020, 338: 135865. |
| [173] | LI Feifei, CHEN Li, LIU Hongmei, et al. Enhanced N2-fixation by engineering the edges of two-dimensional transition-metal disulfides[J]. The Journal of Physical Chemistry C, 2019, 123(36): 22221-22227. |
| [174] | LI Xianghong, LI Tingshuai, MA Yongjun, et al. Boosted electrocatalytic N2 reduction to NH3 by defect-rich MoS2 nanoflower[J]. Advanced Energy Materials, 2018, 8(30): 1801357. |
| [175] | WANG Yanwei, WAN Jin, TIAN Wu, et al. Theoretical screening of VSe2 as support for enhanced electrocatalytic performance of transition-metal single atoms[J]. Journal of Colloid and Interface Science, 2021, 590: 210-218. |
| [176] | CAI Lejuan, ZHANG Ning, QIU Bocheng, et al. Computational design of transition metal single-atom electrocatalysts on PtS2 for efficient nitrogen reduction[J]. ACS Applied Materials & Interfaces, 2020, 12(18): 20448-20455. |
| [177] | BAO Anyu, XU Ying, CAO Yong, et al. Theoretical screening and investigation on electrocatalytic nitrogen fixation of single transition metal atom supported by monolayer SnS2 [J]. Applied Surface Science, 2023, 615: 156362. |
| [178] | YANG Tong, SONG Tingting, ZHOU Jun, et al. High-throughput screening of transition metal single atom catalysts anchored on molybdenum disulfide for nitrogen fixation[J]. Nano Energy, 2020, 68: 104304. |
| [179] | ZHAI Xingwu, LI Lei, LIU Xiaoyue, et al. A DFT screening of single transition atoms supported on MoS2 as highly efficient electrocatalysts for the nitrogen reduction reaction[J]. Nanoscale, 2020, 12(18): 10035-10043. |
| [180] | GUO Haoran, LI Lei, WANG Xingyong, et al. Theoretical investigation on the single transition-metal atom-decorated defective MoS2 for electrocatalytic ammonia synthesis[J]. ACS Applied Materials & Interfaces, 2019, 11(40): 36506-36514. |
| [181] | RAFIQ Madiha, HU Xiaozhen, YE Zhiliang, et al. Recent advances in structural engineering of 2D hexagonal boron nitride electrocatalysts[J]. Nano Energy, 2022, 91: 106661. |
| [182] | YU Linke, LI Fengyu. Pt2 dimer anchored vertically in defective BN monolayer as an efficient catalyst for N2 reduction: A DFT study[J]. Catalysts, 2022, 12(11): 1387. |
| [183] | ZAFARI Mohammad, ANAND Rohit, NISSIMAGOUDAR Arun S, et al. Single-atom catalysts supported on a hybrid structure of boron nitride/graphene for efficient nitrogen fixation via synergistic interfacial interactions[J]. Nanoscale, 2024, 16(2): 555-563. |
| [184] | LIU Zaichun, HUANG Ting, CHANG Huhu, et al. Computational design of single Mo atom anchored defective boron phosphide monolayer as a high-performance electrocatalyst for the nitrogen reduction reaction[J]. Energy & Environmental Materials, 2021, 4(2): 255-262. |
| [185] | Deniz ÇAKıR, KECIK Deniz, SAHIN Hasan, et al. Realization of a p-n junction in a single layer boron-phosphide[J]. Physical Chemistry Chemical Physics, 2015, 17(19): 13013-13020. |
| [186] | JIANG H R, SHYY W, LIU M, et al. Boron phosphide monolayer as a potential anode material for alkali metal-based batteries[J]. Journal of Materials Chemistry A, 2017, 5(2): 672-679. |
| [187] | XIAO Xiao, ZOU Lianli, PANG Huan, et al. Synthesis of micro/nanoscaled metal-organic frameworks and their direct electrochemical applications[J]. Chemical Society Reviews, 2020, 49(1): 301-331. |
| [188] | CUI Qianyi, QIN Gangqiang, WANG Weihua, et al. Mo-based 2D MOF as a highly efficient electrocatalyst for reduction of N2 to NH3: A density functional theory study[J]. Journal of Materials Chemistry A, 2019, 7(24): 14510-14518. |
| [189] | SUN Yongxiu, SHI Wenwu, SUN Mengxuan, et al. Molybdenum based 2D conductive metal-organic frameworks as efficient single-atom electrocatalysts for N2 reduction: A density functional theory study[J]. International Journal of Hydrogen Energy, 2023, 48(52): 19972-19983. |
| [190] | ZOU Lianli, WEI Yongsheng, HOU Chunchao, et al. Single-atom catalysts derived from metal-organic frameworks for electrochemical applications[J]. Small, 2021, 17(16): 2004809. |
| [191] | LIU Yaxin, ZHANG Hong, CHENG Xinlu. Electrocatalytic nitrogen fixation performance of two-dimensional metal-organic frameworks Cu3(C6O6) and TM/Cu3(C6O6) from first-principle study[J]. Chemical Physics, 2023, 568: 111837. |
| [1] | LIU Chao, DING Chengao, WU Baoshun, LEI Xinyu, WANG Guangying, YU Zhengwei. Effect of TiO2 support particle size on the denitrification and water/sulfur poisoning resistance of RuO x -V2O5-WO3/TiO2 catalyst [J]. Chemical Industry and Engineering Progress, 2025, 44(S1): 232-242. |
| [2] | LI Xiang, LI Jiaying, NI Heng, SUN Haoran, CAO Jiawei, CHEN Yuxuan, LIU Fengjiao. Advances in the prediction of activation energy barriers for hydrogen atom transfer reactions [J]. Chemical Industry and Engineering Progress, 2025, 44(6): 3336-3344. |
| [3] | WANG Xiaonan, FU Siwei, LIU Kuan, LIN Congsheng, LIN Xiaofeng. Machine learning methods for sustainable alternatives and transition of energy materials [J]. Chemical Industry and Engineering Progress, 2025, 44(5): 2767-2776. |
| [4] | LI Zhangliang, YANG Yuezhu, WU Chuantian, LYU Yuancai. Degradation of bisphenol A by N-TiO2/MoS2/N-TiO2 immobilized laccase on activated carbon fiber felt [J]. Chemical Industry and Engineering Progress, 2025, 44(2): 887-898. |
| [5] | ZHUANG Ke, CHEN Hong, XU Yun, ZHONG Zhaoping, ZHOU Junwu, ZHOU Kai, DONG Yuehong. Resistance of SiO2 modified Ce-V-W/Ti catalyst support to alkali (earth) metal poisoning [J]. Chemical Industry and Engineering Progress, 2025, 44(1): 266-276. |
| [6] | ZHANG Ridong, LYU Jianhua, LIU Jidong, GUO Bao, LI Wensong. Ru-K-NaY catalyzed decarbonylation of dimethyl oxalate to dimethyl carbonate [J]. Chemical Industry and Engineering Progress, 2024, 43(S1): 382-390. |
| [7] | SONG Caicheng, CHEN Xiaozhen, LIU Li, YANG Chengmin, ZHENG Bumei, YIN Xiaoying, SUN Jin, YAO Yunhai, DUAN Weiyu. Research progress of carbon-based carrier supported hydrodesulfurization catalysts [J]. Chemical Industry and Engineering Progress, 2024, 43(S1): 305-314. |
| [8] | XIONG Lei, DING Feiyan, LI Cong, WANG Qunle, LYU Qi, ZHAI Xiaona, LIU Feng. Recent advances in metal Pt supported heterogeneous catalysts [J]. Chemical Industry and Engineering Progress, 2024, 43(S1): 295-304. |
| [9] | LI Lin, HUANG Guoyong, XU Shengming, YU Fengshan, WENG Yaqing, CAO Caifang, WEN Jiawei, WANG Chunxia, WANG Junlian, GU Bintao, ZHANG Yuanhua, LIU Bin, WANG Caiping, PAN Jianming, XU Zeliang, WANG Chong, WANG Ke. Recovery and regeneration preparation of aluminum-based spent catalyst support [J]. Chemical Industry and Engineering Progress, 2024, 43(S1): 640-649. |
| [10] | YIN Chenyang, LIU Yongfeng, CHEN Ruizhe, ZHANG Lu, SONG Jin’ou, LIU Haifeng. Kinetic simulation of n-hexane pyrolysis reaction based on quantitative calculations [J]. Chemical Industry and Engineering Progress, 2024, 43(8): 4273-4282. |
| [11] | WAN Chengfeng, LI Zhida, ZHANG Chunyue, LU Lu. Highly efficient electrocatalytic water splitting by MXene supported CoP nanorods [J]. Chemical Industry and Engineering Progress, 2024, 43(6): 3232-3239. |
| [12] | LIU Yurong, WANG Xingbao, LI Wenying. Regulation of catalyst acid sites and its effect on the deep hydrogenation performance of anthracene [J]. Chemical Industry and Engineering Progress, 2024, 43(4): 1832-1839. |
| [13] | GAI Hongwei, ZHANG Chenjun, QU Jingying, SUN Huailu, TUO Yongxiao, WANG Bin, JIN Xu, ZHANG Xi, FENG Xiang, CHEN De. Research progress on catalytic dehydrogenation process intensification for liquid organic hydride carrier hydrogen storage [J]. Chemical Industry and Engineering Progress, 2024, 43(1): 164-185. |
| [14] | GONG Pengcheng, YAN Qun, CHEN Jinfu, WEN Junyu, SU Xiaojie. Properties and mechanism of eriochrome black T degradation by carbon nanotube-cobalt ferrite composites activated persulfate [J]. Chemical Industry and Engineering Progress, 2023, 42(7): 3572-3581. |
| [15] | YU Zhiqing, HUANG Wenbin, WANG Xiaohan, DENG Kaixin, WEI Qiang, ZHOU Yasong, JIANG Peng. B-doped Al2O3@C support for CoMo hydrodesulfurization catalyst and their hydrodesulfurization performance [J]. Chemical Industry and Engineering Progress, 2023, 42(7): 3550-3560. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||