Chemical Industry and Engineering Progress ›› 2020, Vol. 39 ›› Issue (1): 137-144.DOI: 10.16085/j.issn.1000-6613.2019-0549
• Energy processes and technology • Previous Articles Next Articles
Solubility of phenolic compounds in low temperature coal tar based on molecular simulation
Guangsheng LI( ),Qiang XIE(),Xianglan ZHANG,Haiyong ZHANG
),Qiang XIE(),Xianglan ZHANG,Haiyong ZHANG
- School of Chemical & Environmental Engineering, China University of Mining & Technology, Beijing 100083, China
-
Received:2019-04-10Online:2020-01-14Published:2020-01-05 -
Contact:Qiang XIE
基于分子模拟的低温煤焦油中酚类化合物的溶解特性
李光升(),解强(),张香兰,张海永
- 中国矿业大学(北京)化学与环境工程学院,北京 100083
-
通讯作者:解强 -
作者简介:李光升(1996—),男,博士研究生,研究方向为煤基液体产物的精细化分离。E-mail:kdlgsheng@163.com 。 -
基金资助:国家重点研发计划(2016YFB0600305-2)
CLC Number:
Cite this article
Guangsheng LI,Qiang XIE,Xianglan ZHANG,Haiyong ZHANG. Solubility of phenolic compounds in low temperature coal tar based on molecular simulation[J]. Chemical Industry and Engineering Progress, 2020, 39(1): 137-144.
李光升,解强,张香兰,张海永. 基于分子模拟的低温煤焦油中酚类化合物的溶解特性[J]. 化工进展, 2020, 39(1): 137-144.
share this article
Add to citation manager EndNote|Ris|BibTeX
URL: https://hgjz.cip.com.cn/EN/10.16085/j.issn.1000-6613.2019-0549
| 组分 | 结构差异 | 典型化合物 |
|---|---|---|
| 酚类化合物 | 烷基取代基碳数 | 苯酚、邻甲基苯酚、邻乙基苯酚、邻丙基苯酚、邻叔丁基苯酚 |
| 烷基取代基数量 | 苯酚、邻甲基苯酚、2,3-二甲基苯酚 | |
| 烷基取代基位置 | 邻甲酚、间甲酚、对甲酚 | |
| 羟基数量 | 苯酚、邻苯二酚 | |
| 羟基位置 | 邻苯二酚、间苯二酚、对苯二酚 | |
| 芳香烃 | 烷基取代基碳数 | 苯、甲苯、乙苯、异丙苯 |
| 烷基取代基数量 | 苯、甲苯、邻二甲苯 | |
| 烷基取代基位置 | 邻二甲苯、间二甲苯、对二甲苯 | |
| 苯环数量 | 苯、萘、蒽 | |
| 脂肪烃 | 直链烃碳链长短 | 正己烷、正十二烷、正二十二烷 |
| 环状烃与直链烃 | 正己烷、环戊烷、环己烷 | |
| 杂环氮化物 | 苯环数量等 | 吡啶、喹啉、吲哚、咔唑 |
| 含硫化合物 | 苯环数量等 | 噻吩、苯并噻吩、二苯并噻吩 |
| 组分 | 结构差异 | 典型化合物 |
|---|---|---|
| 酚类化合物 | 烷基取代基碳数 | 苯酚、邻甲基苯酚、邻乙基苯酚、邻丙基苯酚、邻叔丁基苯酚 |
| 烷基取代基数量 | 苯酚、邻甲基苯酚、2,3-二甲基苯酚 | |
| 烷基取代基位置 | 邻甲酚、间甲酚、对甲酚 | |
| 羟基数量 | 苯酚、邻苯二酚 | |
| 羟基位置 | 邻苯二酚、间苯二酚、对苯二酚 | |
| 芳香烃 | 烷基取代基碳数 | 苯、甲苯、乙苯、异丙苯 |
| 烷基取代基数量 | 苯、甲苯、邻二甲苯 | |
| 烷基取代基位置 | 邻二甲苯、间二甲苯、对二甲苯 | |
| 苯环数量 | 苯、萘、蒽 | |
| 脂肪烃 | 直链烃碳链长短 | 正己烷、正十二烷、正二十二烷 |
| 环状烃与直链烃 | 正己烷、环戊烷、环己烷 | |
| 杂环氮化物 | 苯环数量等 | 吡啶、喹啉、吲哚、咔唑 |
| 含硫化合物 | 苯环数量等 | 噻吩、苯并噻吩、二苯并噻吩 |
| 物质 | 分子数量 | 质量分数/% |
|---|---|---|
| 苯酚 | 1331 | 45.1 |
| 苯 | 1080 | 30.4 |
| 正己烷 | 500 | 15.5 |
| 吲哚 | 143 | 6 |
| 苯并噻吩 | 63 | 3 |
| 物质 | 分子数量 | 质量分数/% |
|---|---|---|
| 苯酚 | 1331 | 45.1 |
| 苯 | 1080 | 30.4 |
| 正己烷 | 500 | 15.5 |
| 吲哚 | 143 | 6 |
| 苯并噻吩 | 63 | 3 |

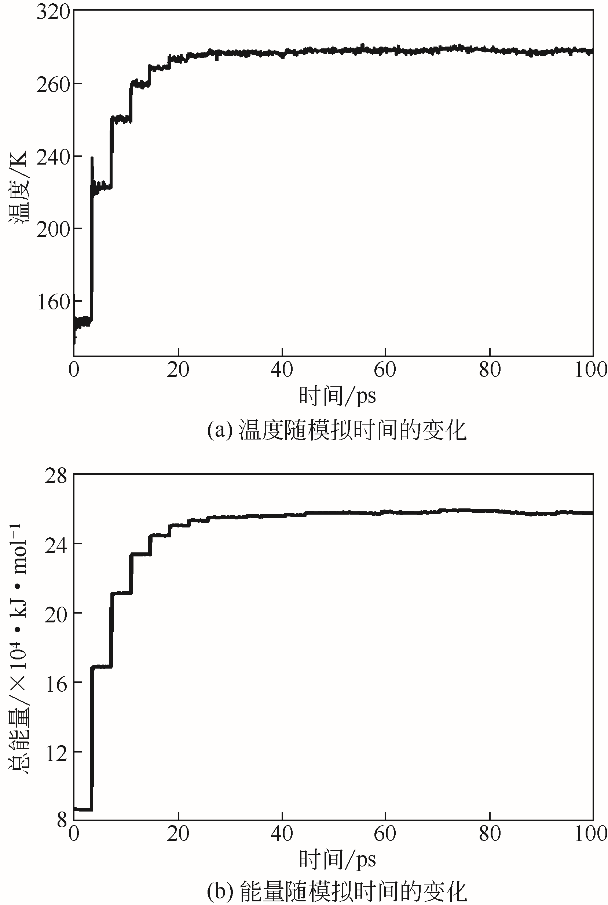
| 物质 | 分子数量 | 密度①/g·cm-3 | 密度②/g·cm-3 |
|---|---|---|---|
| 苯酚 | 200 | 1.083 | 1.0576 |
| 苯 | 200 | 0.863 | 0.8737 |
| 正己烷 | 200 | 0.661 | 0.6603 |
| 吲哚 | 200 | 1.098 | 1.22 |
| 苯并噻吩 | 200 | 1.130 | 1.165 |
| 物质 | 分子数量 | 密度①/g·cm-3 | 密度②/g·cm-3 |
|---|---|---|---|
| 苯酚 | 200 | 1.083 | 1.0576 |
| 苯 | 200 | 0.863 | 0.8737 |
| 正己烷 | 200 | 0.661 | 0.6603 |
| 吲哚 | 200 | 1.098 | 1.22 |
| 苯并噻吩 | 200 | 1.130 | 1.165 |
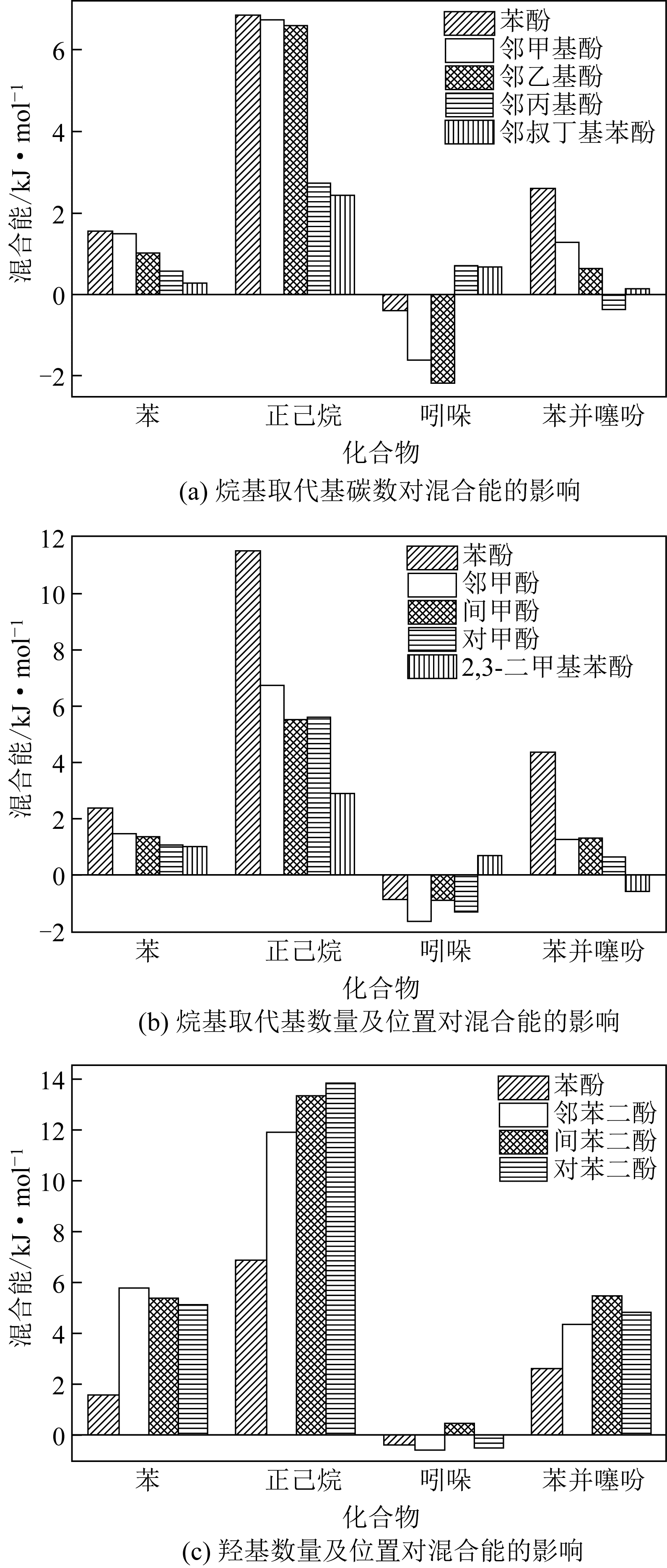
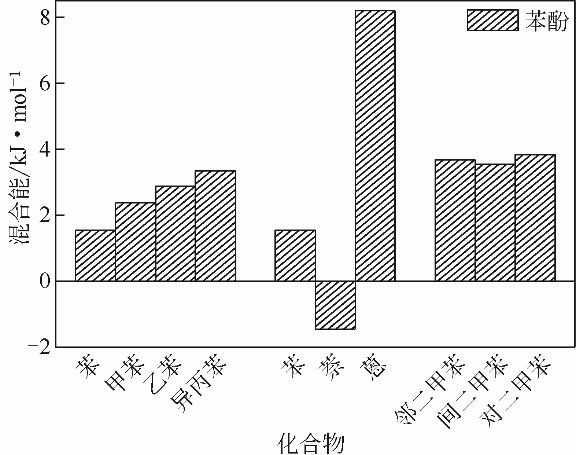
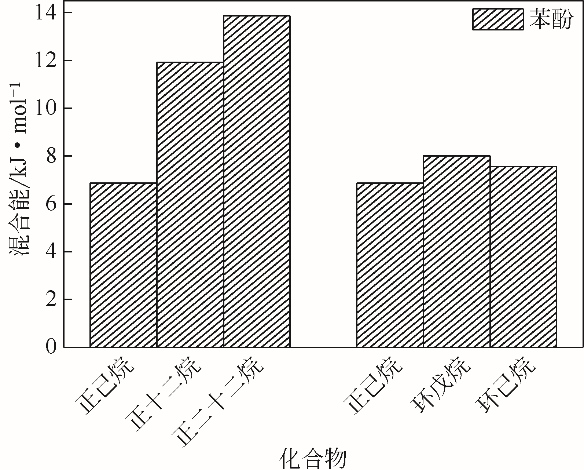
| 物质 | 混合能/kJ·mol-1 |
|---|---|
| 苯酚-吲哚 | -0.396 |
| 苯酚-喹啉 | -1.357 |
| 苯酚-吡啶 | -5.745 |
| 苯酚-咔唑 | 12.641 |
| 物质 | 混合能/kJ·mol-1 |
|---|---|
| 苯酚-吲哚 | -0.396 |
| 苯酚-喹啉 | -1.357 |
| 苯酚-吡啶 | -5.745 |
| 苯酚-咔唑 | 12.641 |
| 物质 | 混合能/kJ·mol-1 |
|---|---|
| 苯酚-噻吩 | 5.303 |
| 苯酚-苯并噻吩 | 2.535 |
| 苯酚-二苯并噻吩 | 24.694 |
| 物质 | 混合能/kJ·mol-1 |
|---|---|
| 苯酚-噻吩 | 5.303 |
| 苯酚-苯并噻吩 | 2.535 |
| 苯酚-二苯并噻吩 | 24.694 |
| 物质 | Einter/×103 kJ·mol-1 |
|---|---|
| 苯酚-苯 | -34.86 |
| 苯酚-正己烷 | -13.57 |
| 苯酚-吲哚 | -7.00 |
| 苯酚-苯并噻吩 | -2.26 |
| 物质 | Einter/×103 kJ·mol-1 |
|---|---|
| 苯酚-苯 | -34.86 |
| 苯酚-正己烷 | -13.57 |
| 苯酚-吲哚 | -7.00 |
| 苯酚-苯并噻吩 | -2.26 |
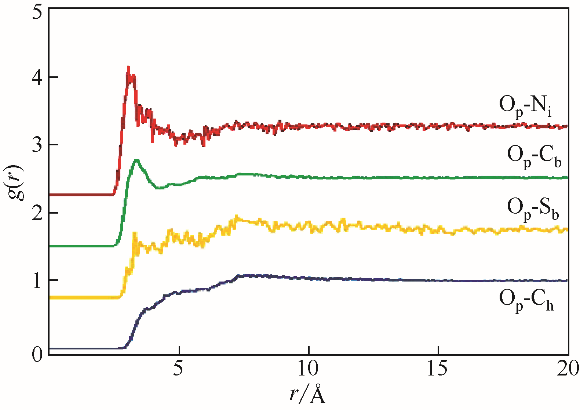
| 1 | 何璐, 解强, 梁鼎成, 等. 煤热解反应热测定方法研究进展[J]. 化工进展, 2017, 36(2): 494-501. |
| HE L, XIE Q, LIANG D C,et al. Measurement of reaction heat of coal pyrolysis: state-of-the-art[J]. Chemical Industry and Engineering Progress, 2017, 36(2): 494-501. | |
| 2 | 张俊杰, 徐绍平, 王光永, 等. 停留时间对低阶煤快速热解产物分布、组成及结构的影响[J]. 化工进展, 2019, 38(3): 1346-1352. |
| ZHANG J J, XU S P, WANG G Y, et al. Effect of residence time on distribution, composition and structure of products derived from fast coal pyrolysis[J]. Chemical Industry and Engineering Progress, 2019, 38(3): 1346-1352. | |
| 3 | SUN M, CHEN J, DAI X M, et al. Controlled separation of low temperature coal tar based on solvent extraction-column chromatography[J]. Fuel Processing Technology, 2015, 136: 41-49. |
| 4 | 张生娟, 高亚男, 陈刚, 等. 煤焦油中酚类化合物的分离及其组成结构鉴定研究进展[J]. 化工进展, 2018, 37(7): 2588-2596. |
| ZHANG S J, GAO Y N, CHEN G, et al. Progress of separation, composition and structure identification of phenolic compounds in coal tar[J]. Chemical Industry and Engineering Progress, 2018, 37(7): 2588-2596. | |
| 5 | MENG H, GE C T, REN N N, et al. Complex extraction of phenol and cresol from model coal tar with polyols, ethanol amines, and ionic liquids thereof[J]. Industrial & Engineering Chemistry Research, 2013, 53(1): 355-362. |
| 6 | SUN M, MA X X, YAO Q X, et al. GC-MS and TG-FTIR study of petroleum ether extract and residue from low temperature coal tar[J]. Energy & Fuels, 2011, 25(3): 1140-1145. |
| 7 | SHI Q, PAN N, LONG H Y. Characterization of middle-temperature gasification coal tar. Part 3: Molecular composition of acidic compounds[J]. Energy & Fuels, 2013, 27(1): 108-117. |
| 8 | 李文英, 慕海, 王伟, 等. 煤基粗油轻质组分定性定量分析现状与展望[J]. 化工进展, 2019, 38(1): 217-228. |
| LI W Y, MU H, WANG W, et al. Status quo and outlook of qualitative and quantitative analysis of light weight fractions of coal-based crude oil[J]. Chemical Industry and Engineering Progress, 2019, 38(1): 217-228. | |
| 9 | DAI F F, XIN K, SONG Y H, et al. Liquid-liquid equilibria for the extraction of phenols from alkane using ethylene glycol[J]. Fluid Phase Equilibria, 2016, 419: 50-56. |
| 10 | JIAO T T, ZHUANG X L, HE H Y, et al. Separation of phenolic compounds from coal tar via liquid-liquid extraction using amide compounds[J]. Industrial & Engineering Chemistry Research, 2015, 54(9): 2573-2579. |
| 11 | 易兰, 李文英, 冯杰, 等. 煤基液体油分离技术研究进展[J]. 化工学报, 2017, 68(10): 3678-3692. |
| YI L, LI W Y, FENG J, et al. Recent progress on coal-based liquid oil separation technology[J]. CIESC Journal, 2017, 68(10): 3678-3692. | |
| 12 | 刘兴坤, 张香兰, 刘潜, 等. 正十二烷-甲苯-苯酚三元体系液液相平衡数据的测定与关联[J]. 高校化学工程学报, 2018, 32(2): 275-279. |
| LIU X K, ZHANG X L, LIU Q, et al. Determination and correlation of liquid equilibrium of the n-dodecane-toluene-phenol ternary system[J]. Journal of Chemical Engineering of Chinese Universities, 2018, 32(2): 275-279. | |
| 13 | 张海永, 刘潜, 刘兴坤, 等. 低温煤焦油中正十二烷-甲苯-苯酚的相平衡及分离[J]. 化工学报, 2018, 69(8): 3479-3487. |
| ZHANG H Y, LIU Q, LIU X K, et al. Phase equilibrium and separation of n-dodecane-toluene-phenol in low temperature coal tar[J]. CIESC Journal, 2018, 69(8): 3479-3487. | |
| 14 | 李洪, 张季, 李鑫钢, 等. 分子模拟方法计算相平衡热力学性质的研究进展[J]. 化工进展, 2017, 36(8): 2731-2741. |
| LI H, ZHANG J, LI X G, et al. Progress in study on thermodynamic properties of phase equilibria using molecular simulation[J]. Chemical Industry and Engineering Progress, 2017, 36(8): 2731-2741. | |
| 15 | ZHAO Y, ZHANG X H, ZHANG W, et al. Simulation and experimental on the solvation interaction between the GAP matrix and insensitive energetic plasticizers in solid propellants[J]. The Journal of Physical Chemistry A, 2015, 120(5): 765-770. |
| 16 | FU X L, FAN X Z, JU X H, et al. Molecular dynamic simulations on the interaction between an HTPE polymer and energetic plasticizers in a solid propellant[J]. RSC Advances, 2015, 65(5): 52844-52851. |
| 17 | ZENG F L, SUN Y, ZHOU Y, et al. Molecular simulations of the miscibility in binary mixtures of PVDF and POSS compounds[J]. Modelling and Simulation in Materials Science and Engineering, 2009, 17(7): 75002. |
| 18 | 于共奇. 重质油组分及其溶解机理的分子动力学研究[D]. 青岛: 中国石油大学(华东), 2013. |
| YU G Q. Molecular dynamics study of the composition and the dissolution theory of heavy oil[D]. Qingdao: China University of Petroleum (East China), 2013. | |
| 19 | FAN C F, OLAFSON B D, BLANCO M, et al. Application of molecular simulation to derive phase diagrams of binary mixtures[J]. Macromolecules, 1992, 25(14): 3667-3676. |
| 20 | BLANCO M. Molecular silverware. Ⅰ. General solutions to excluded volume constrained problems[J]. Journal of Computational Chemistry, 1991, 12(2): 237-247. |
| 21 | FLORY. Principles of polymer chemistry[M]. New York: Cornell University Press, 1953: 672. |
| 22 | 周公度. 结构化学基础[M]. 北京: 北京大学出版社, 2008: 363. |
| ZHOU G D. Structural chemistry foundation[M]. Beijing: Peking University Press, 2008: 363. | |
| 23 | 邓蕾, 张炜, 鲍桐, 等. PBT与含能增塑剂相互作用的分子动力学模拟[J]. 含能材料, 2017, 25(1): 32-38. |
| DENG L, ZHANG W, BAO T, et al. Molecular dynamics simulation of interaction between PBT and energetic plasticizer[J]. Chinese Journal of Energetic Materials, 2017, 25(1): 32-38. | |
| 24 | STEINER T, DESIRAJU G R. Distinction between the weak hydrogen bond and the van der Waals interaction[J]. Chemical Communications, 1998(8): 891-892. |
| 25 | PAN N, CUI D C, LI R L, et al. Characterization of middle-temperature gasification coal tar. Part 1: bulk properties and molecular compositions of distillates and basic fractions[J]. Energy & Fuels, 2012, 26(9): 5719-5728. |
| 26 | LONG H Y, SHI Q, PAN N, et al. Characterization of middle-temperature gasification coal tar. Part 2: Neutral fraction by extrography followed by gas chromatography-mass spectrometry and electrospray ionization coupled with fourier transform ion cyclotron resonance mass spectrometry[J]. Energy & Fuels, 2012, 26(6): 3424-3431. |
| 27 | SUN H. COMPASS: an ab initio force-field optimized for condensed-phase applications-overview with details on alkane and benzene compounds[J]. The Journal of Physical Chemistry B, 1988, 102(38): 7338-7364. |
| 28 | 肖瑞华. 煤焦油化工学[M]. 2版. 北京: 冶金工业出版社, 2009: 375-442. |
| XIAO R H. Coal tar chemical engineering[M]. 2nd ed. Beijing: Metallurgical Industry Press, 2009: 375-442. | |
| 29 | ZHANG L Z, ZHANG M, GAO J, et al. Efficient extraction of neutral heterocyclic nitrogen compounds from coal tar via ionic liquids and its mechanism analysis[J]. Energy & Fuels, 2018, 32(9): 9358-9370. |
| [1] | CUI Shoucheng, XU Hongbo, PENG Nan. Simulation analysis of two MOFs materials for O2/He adsorption separation [J]. Chemical Industry and Engineering Progress, 2023, 42(S1): 382-390. |
| [2] | OUYANG Sufang, ZHOU Daowei, HUANG Wei, JIA Feng. Research progress on novel anti-migration rubber antioxidants [J]. Chemical Industry and Engineering Progress, 2023, 42(7): 3708-3719. |
| [3] | LI Ruidong, HUANG Hui, TONG Guohu, WANG Yueshe. Hygroscopic properties and corrosion behavior of ammonium salt in a crude oil distillation column [J]. Chemical Industry and Engineering Progress, 2023, 42(6): 2809-2818. |
| [4] | ZHAO Yi, YANG Zhen, ZHANG Xinwei, WANG Gang, YANG Xuan. Molecular simulation of self-healing behavior of asphalt under different crack damage and healing temperature [J]. Chemical Industry and Engineering Progress, 2023, 42(6): 3147-3156. |
| [5] | YANG Farong, GU Lili, LIU Yang, LI Weixue, CAI Jieyun, WANG Huiping. Preparation and application of molecularly imprinted polymers of terbutylazine assisted by computer simulation [J]. Chemical Industry and Engineering Progress, 2023, 42(6): 3157-3166. |
| [6] | SANG Wei, TANG Jianfeng, HUA Yihuai, CHEN Jie, SUN Peiyuan, XU Yifei. Effects of physical solvent and amine properties on the performance of biphasic solvent [J]. Chemical Industry and Engineering Progress, 2023, 42(4): 2151-2159. |
| [7] | SONG Chao, YE Xuemin, LI Chunxi. Molecular dynamics study on the influence of self-assembly behaviors of nanoparticles and surfactants on the properties of silicone oil/water interface [J]. Chemical Industry and Engineering Progress, 2022, 41(S1): 366-375. |
| [8] | ZHU Hao, LIU Hanfei, JI Yufan, LI Shuangtao, HUANG Yiping, GAO Yuan, WEI Zhenhao, ZHU Kai, HAN Weiqing, WEI Kajia. Research advance and mechanism analysis of catalytic ozonation of phenolic compounds [J]. Chemical Industry and Engineering Progress, 2022, 41(S1): 545-555. |
| [9] | LI Yanping, YAN Dazhou, YANG Tao, WEN Guosheng, HAN Zhicheng. Removal of methylchlorosilane in silicon-based electron gas by molecular dynamics simulation [J]. Chemical Industry and Engineering Progress, 2022, 41(8): 4375-4385. |
| [10] | FENG Ying, ZHAO Mengjie, CUI Qian, XIE Yuju, ZHANG Jianwei, DONG Xin. Research progress of molecular simulation technology in the development and application of chitosan functional materials [J]. Chemical Industry and Engineering Progress, 2022, 41(8): 4241-4253. |
| [11] | ZHANG Xincheng, HE Lin, SUI Hong, LI Xingang. Demulsification process and enhancement by viscosity reduction for water-in-heavy oil emulsions [J]. Chemical Industry and Engineering Progress, 2022, 41(7): 3534-3544. |
| [12] | MA Xu, ZOU Minggui, CUI Weiwei, FU Anran, LIAO Xiaolong, GONG Guifen. Synthesis and performance of a kind of water soluble negative electrode binder [J]. Chemical Industry and Engineering Progress, 2022, 41(6): 3138-3145. |
| [13] | CHEN Lei, YAN Xingqing, HU Yanwei, YU Shuai, YANG Kai, CHEN Shaoyun, GUAN Hui, YU Jianliang, MAHGEREFTEH Haroun, MARTYNOV Sergey. Research progress on fracture control of accidental leakage and decompression in CO2 pipeline transportation [J]. Chemical Industry and Engineering Progress, 2022, 41(3): 1241-1255. |
| [14] | GU Zhipan, YANG Jichun, ZHANG Ye, TAO Leren, LIU Fanhan. Mathematical modelling of water sorption isotherms and thermodynamic properties of municipal sewage sludge [J]. Chemical Industry and Engineering Progress, 2022, 41(2): 998-1008. |
| [15] | QIN Li, LI Dongdong, TIAN Jingsheng, HAN Junhua, ZHANG Yin, LI Jianwen, GAO Wenhui. Preparation of leuco malachite green imprinting sensor based on multiple technologies and its application [J]. Chemical Industry and Engineering Progress, 2022, 41(11): 6018-6028. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||