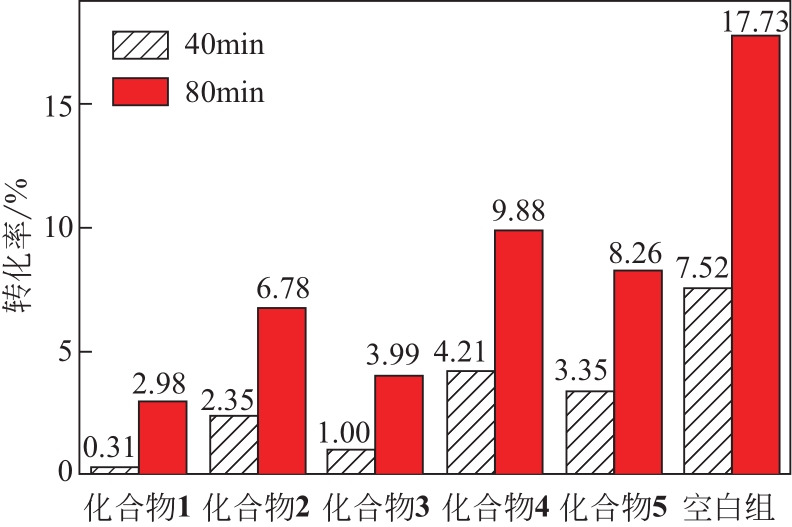
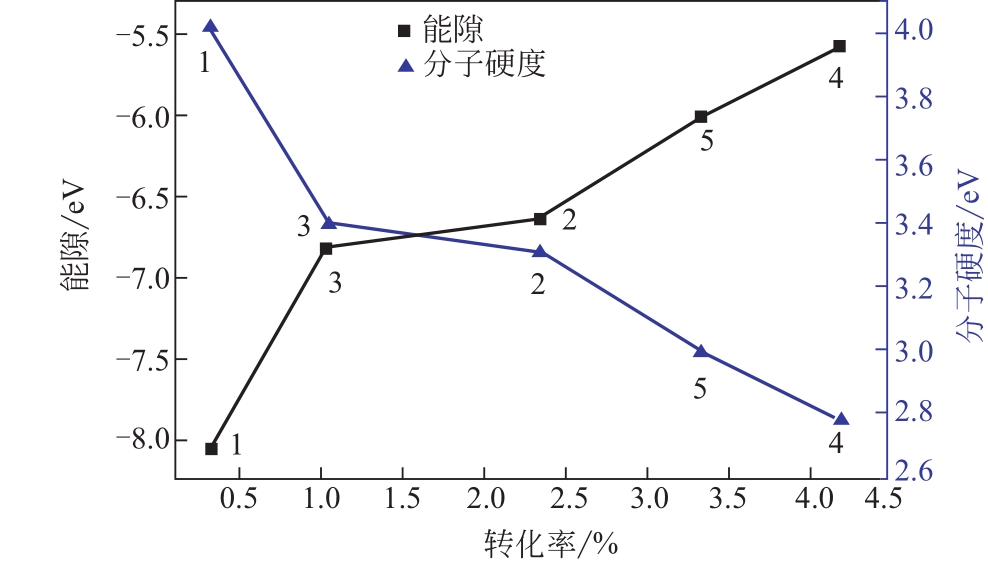
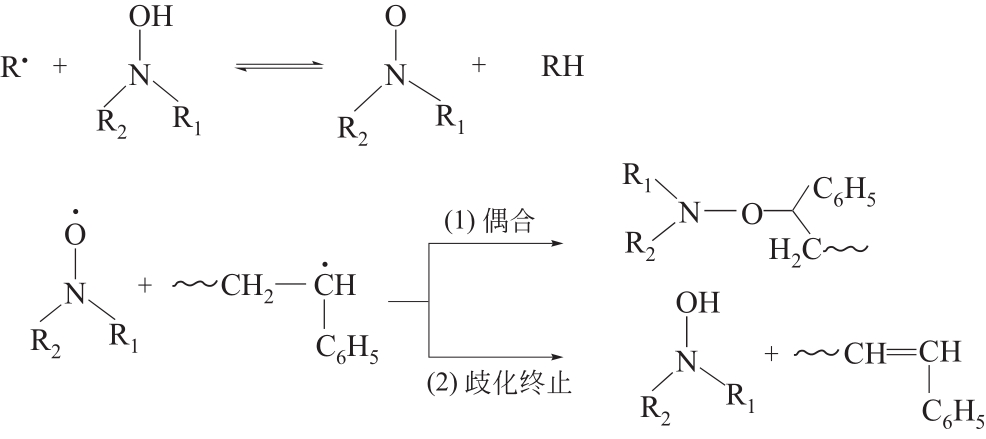
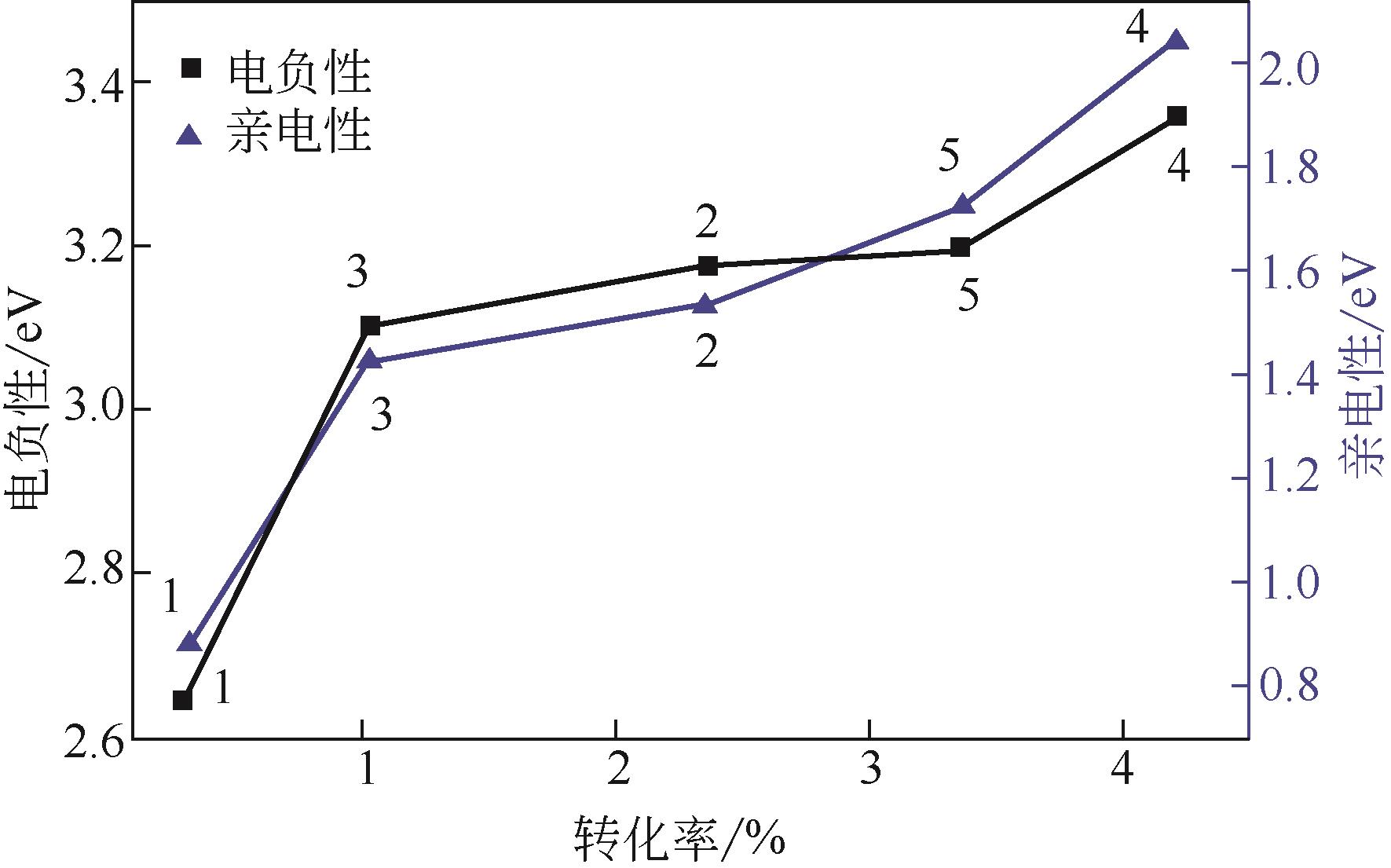
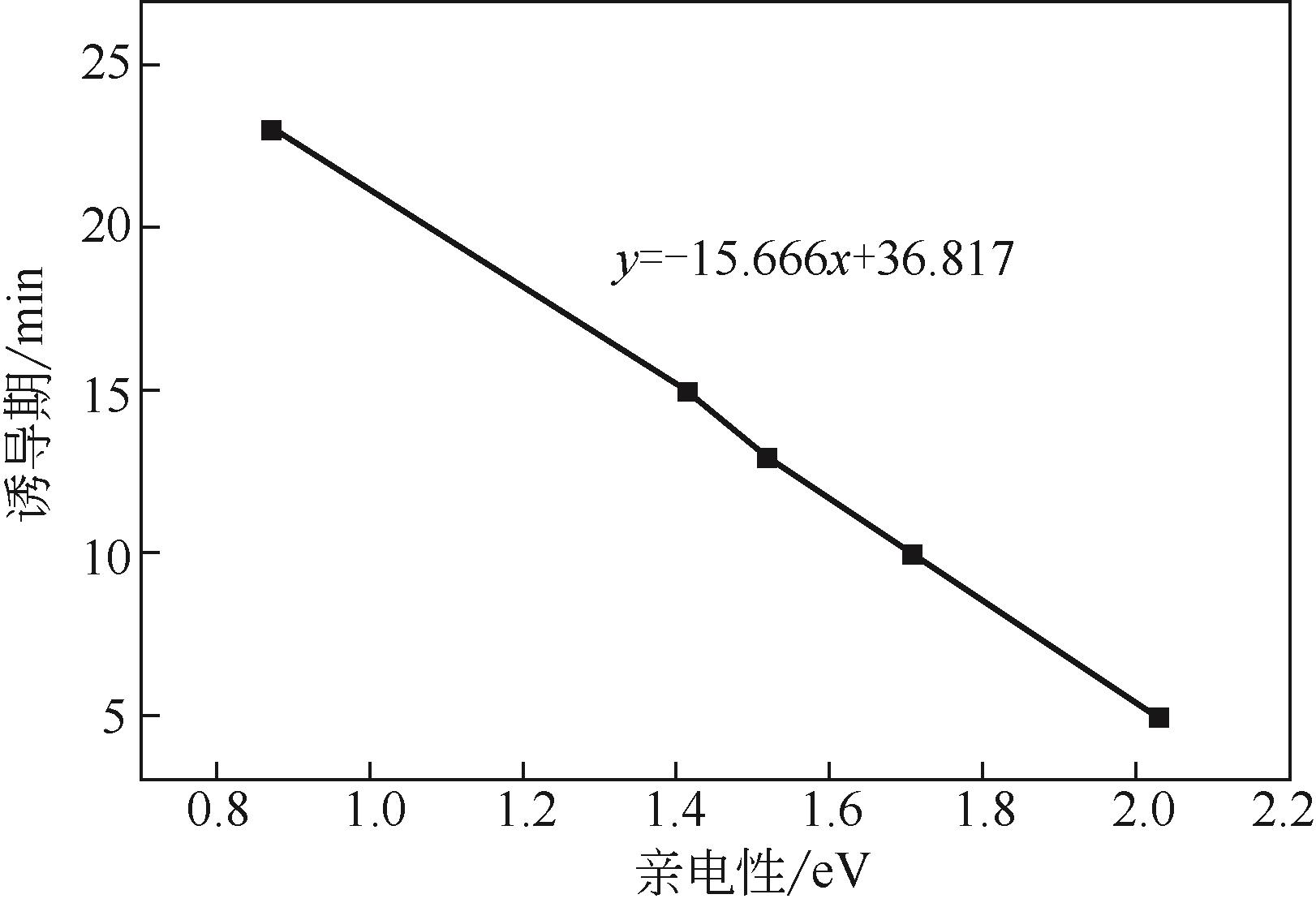
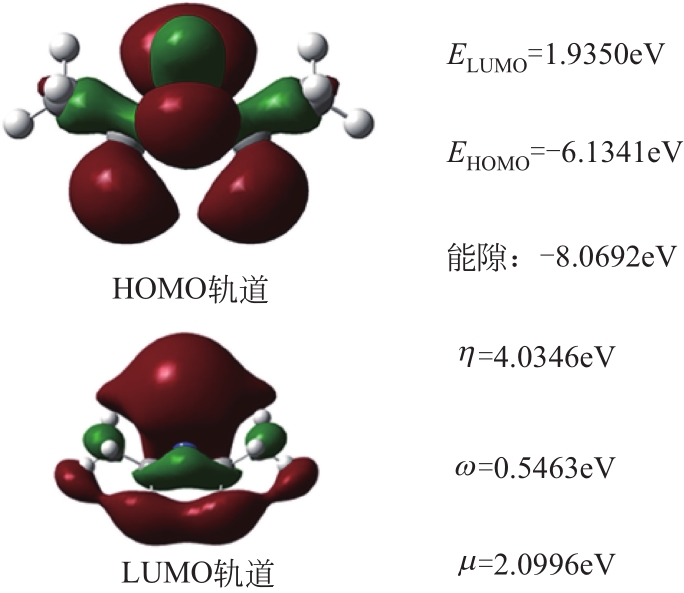
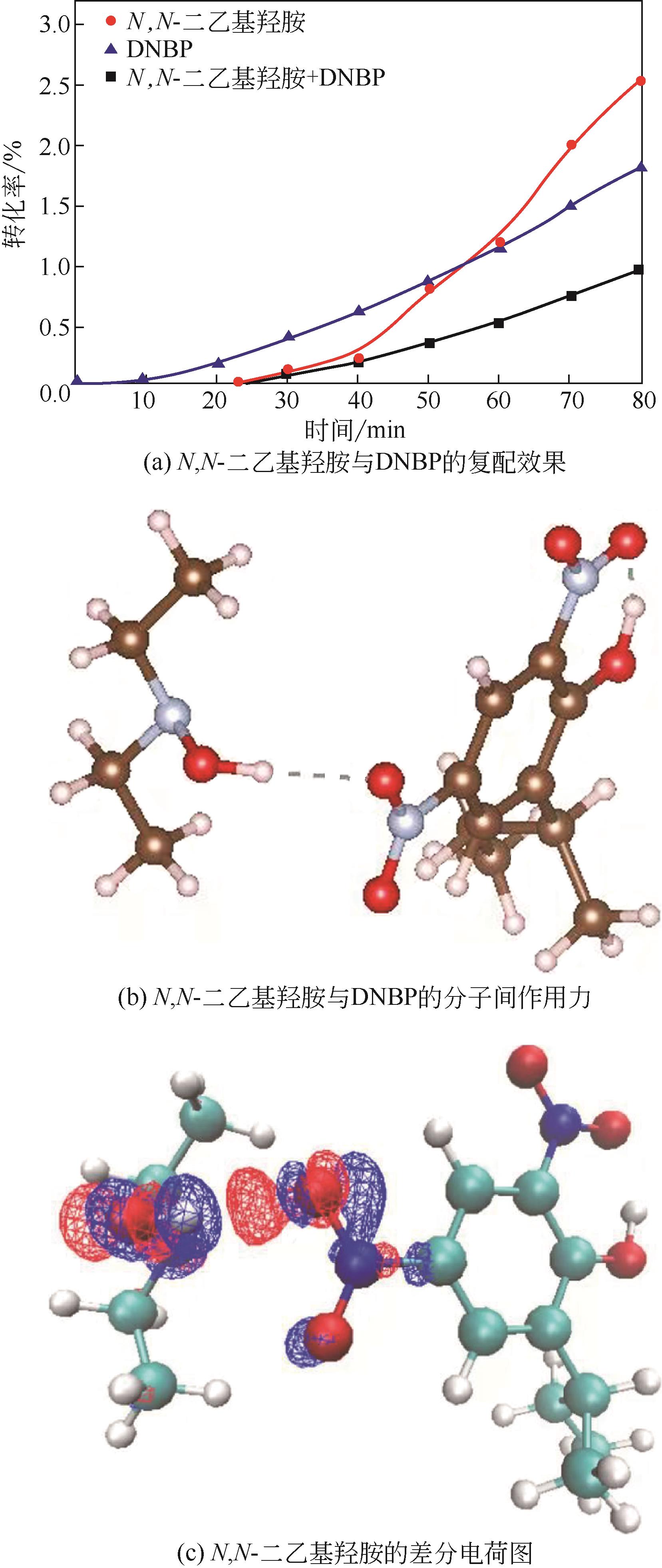
| 1 |
GHOSH J, GHORAI S, BHUNIA S, et al. The role of devulcanizing agent for mechanochemical devulcanization of styrene butadiene rubber vulcanizate[J]. Polymer Engineering & Science, 2018, 58(1): 74-85.
|
| 2 |
PULYALINA A Y, ROSTOVTSEVA V A, PIENTKA Z, et al. Hybrid gas separation membranes containing star-shaped polystyrene with the fullerene (C60) core[J]. Petroleum Chemistry, 2018, 58(4): 296-303.
|
| 3 |
CAI S, ZHANG B, CREMASCHI L. Review of moisture behavior and thermal performance of polystyrene insulation in building applications[J]. Building and Environment, 2017, 123: 50-65.
|
| 4 |
DIMIAN A C, BILDEA C S. Energy efficient styrene process: design and plantwide control[J]. Industrial & Engineering Chemistry Research, 2019, 58(12): 4890-4905.
|
| 5 |
ZHAO L, ZHU W, PAPADAKI M I, et al. Probing into styrene polymerization runaway hazards: effects of the monomer mass fraction[J]. ACS Omega, 2019, 4(5): 8136-8145.
|
| 6 |
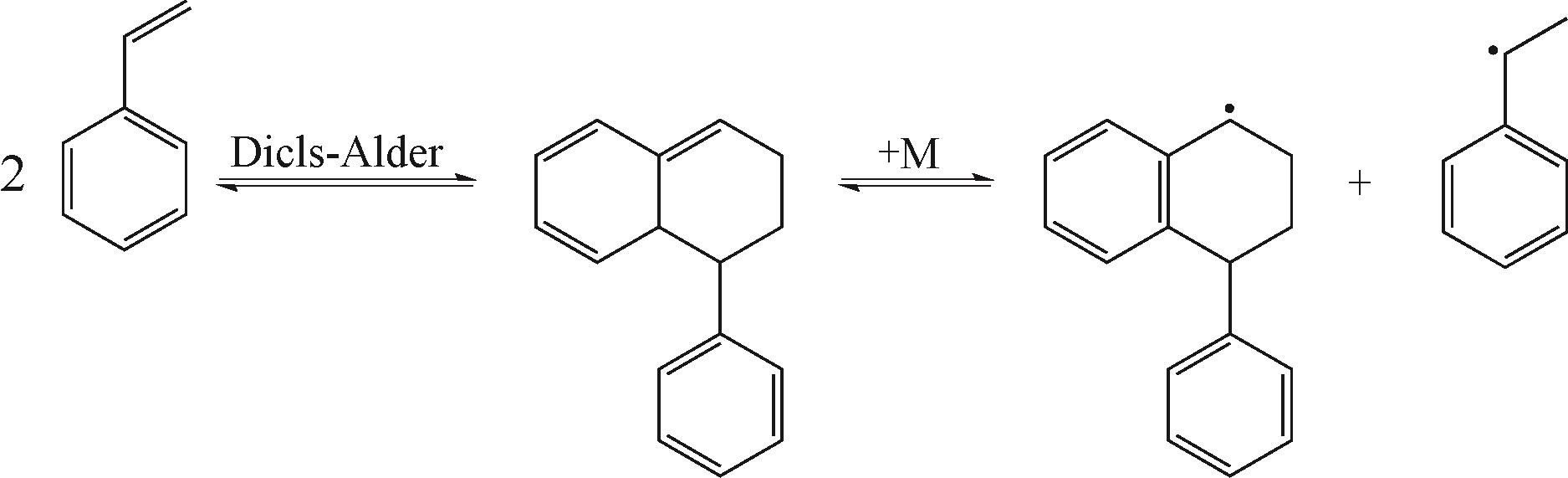
MAYO F R. The dimerization of styrene[J]. Journal of the American Chemical Society, 1968, 90(5): 1289-1295.
|
| 7 |
齐向伟. 新型真阻聚剂在苯乙烯装置的应用[J]. 化工设计通讯, 2018, 44(5): 78, 80.
|
|
QI Xiangwei. Application of new true polymerization inhibitor in styrene unit[J]. Chemical Engineering Design Communications, 2018, 44(5): 78, 80.
|
| 8 |
练成, 刘洪来. 经典密度泛函理论在双电层超级电容器研究中的应用[J]. 化工进展, 2019, 38(1): 244-260.
|
|
LIAN Cheng, LIU Honglai. Classic density functional theory for designing supercapacitors[J]. Chemical Industry and Engineering Progress, 2019, 38(1): 244-260.
|
| 9 |
李南, 马国强, 车海英, 等. 密度泛函理论在高电压电解液设计中的应用[J]. 化工进展, 2019, 38(7): 3253-3264.
|
|
LI Nan, MA Guoqiang, CHE Haiying, et al. Application of density functional theory in the design of high potential electrolyte[J]. Chemical Industry and Engineering Progress, 2019, 38(7): 3253-3264.
|
| 10 |
ZHENG Y Z, CHEN D F, DENG G, et al. The substituent effect on the radical scavenging activity of apigenin[J]. Molecules, 2018, 23(8): 1989-1999.
|
| 11 |
DE VLEESCHOUWER F, SPEYBROECK V VAN, WAROQUIER M, et al. Electrophilicity and nucleophilicity index for radicals[J]. Organic Letters, 2007, 9(14): 2721-2724.
|
| 12 |
MCGARRY L W. Method of making hydroxy-substituted hydroxylamines and color developers containing same: US6028225(A)[P]. 2000-02-22.
|
| 13 |
SELTZER R, DEVORE D, CUNKLE G T, et al. Inhibition of pulp and paper yellowing using hydroxylamines and other coadditives: US2002/88574[P]. 2002-07-11.
|
| 14 |
BALDWIN J E, HARWOOD L M, LOMBARD M J. A general procedure for the synthesis of isoxazolidin-5-ones[J]. Tetrahedron, 1984, 40(21): 4363-4370.
|
| 15 |
LEAVER S A, PALANIANDAVAR M, KILNER C A, et al. A new synthesis of bis(2-{pyrid-2-yl}ethyl)amine (LH) from bis(2-{pyrid-2-yl}ethyl)hydroxylamine (LOH), and the copper-dependent reduction of LOH to LH[J]. Dalton Transactions, 2003, 22(22): 4224-4225.
|
| 16 |
杜飞, 马龙, 郭澄, 等. N,N-二苄基羟胺的清洁化合成工艺研究[J]. 精细化工, 2013, 30(7): 781-783.
|
|
DU Fei, MA Long, GUO Cheng, et al. A study on the clean synthetic technology of N,N-dibenzyl hydroxylamine[J]. Fine Chemicals, 2013, 30(7): 781-783.
|
| 17 |
MAILLARD B, INGOLD K U, SCAIANO J C. Rate constants for the reactions of free radicals with oxygen in solution[J]. Journal of the American Chemical Society, 1983, 105(15): 5095-5099.
|
| 18 |
GOGOTOV A F. High-performance thermal polymerization inhibitors based on 4-tert-butylcatechol and its compositions for petrochemical plants[J]. Petroleum Chemistry, 2017, 57(10): 891-896.
|
| 19 |
AL-MAJEDY Y K, AL-DUHAIDAHAWI D L, AL-AZAWI K F, et al. Coumarins as potential antioxidant agents complemented with suggested mechanisms and approved by molecular modeling studies[J]. Molecules, 2016, 21(2): 135-145.
|
| 20 |
汪汉卿, 冯良波, 蔡邦华, 等. 羟胺类化合物在烯烃单体自由基聚合反应中形成氮氧自由基的ESR研究[J]. 波普学杂志,1989, 6(3): 369-376.
|
|
WANG Hanqin, FENG Liangbo, CAI Banghua, et al. An ESR study of nitroxide radicals produced in the radical polymerization of vinyl monomer[J]. Chinese Journal of Magnetic Resonance, 1989, 6(3): 369-376.
|
| 21 |
李雪云. 苯乙烯精馏阻聚剂的研究[D]. 上海: 华东理工大学, 2011.
|
|
LI Xueyun. Study on the inhibitor in the styrene distillation processing[D]. Shanghai: East China University of Science and Technology, 2011.
|
| 22 |
张自义, 李兆陇, 王晓艳, 等. 乙烯系单体自由基聚合的阻聚效应 Ⅹ.羟胺类化合物对苯乙烯自由基聚合的阻聚及历程[J]. 高分子学报, 1990(2): 233-238.
|
|
ZHANG Ziyi, LI Zhaolong, WANG Xiaoyan, et al. Inhibition effect of vinyl monomers in radical polymerization Ⅹ. Inhibition of hydroxylamine compounds on radical polymerization of styrene and its mechanism[J]. Acta Polymerica Sinica, 1990(2): 233-238.
|
 ), 黄占凯2, 赵甲2, 何艳贞1, 赵福利2, 张春丽2, 刘红光2, 韩恩山1(
), 黄占凯2, 赵甲2, 何艳贞1, 赵福利2, 张春丽2, 刘红光2, 韩恩山1(