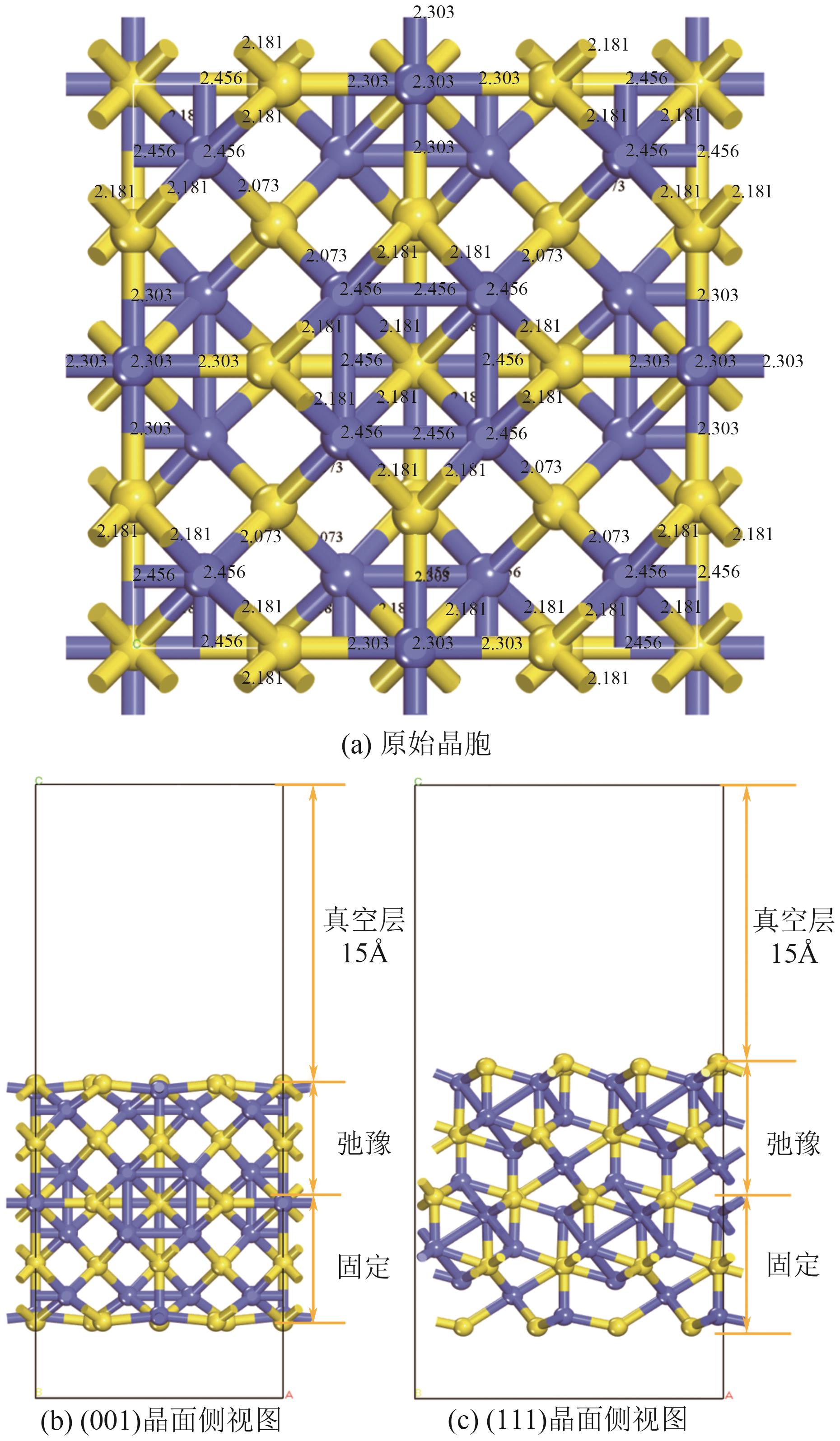
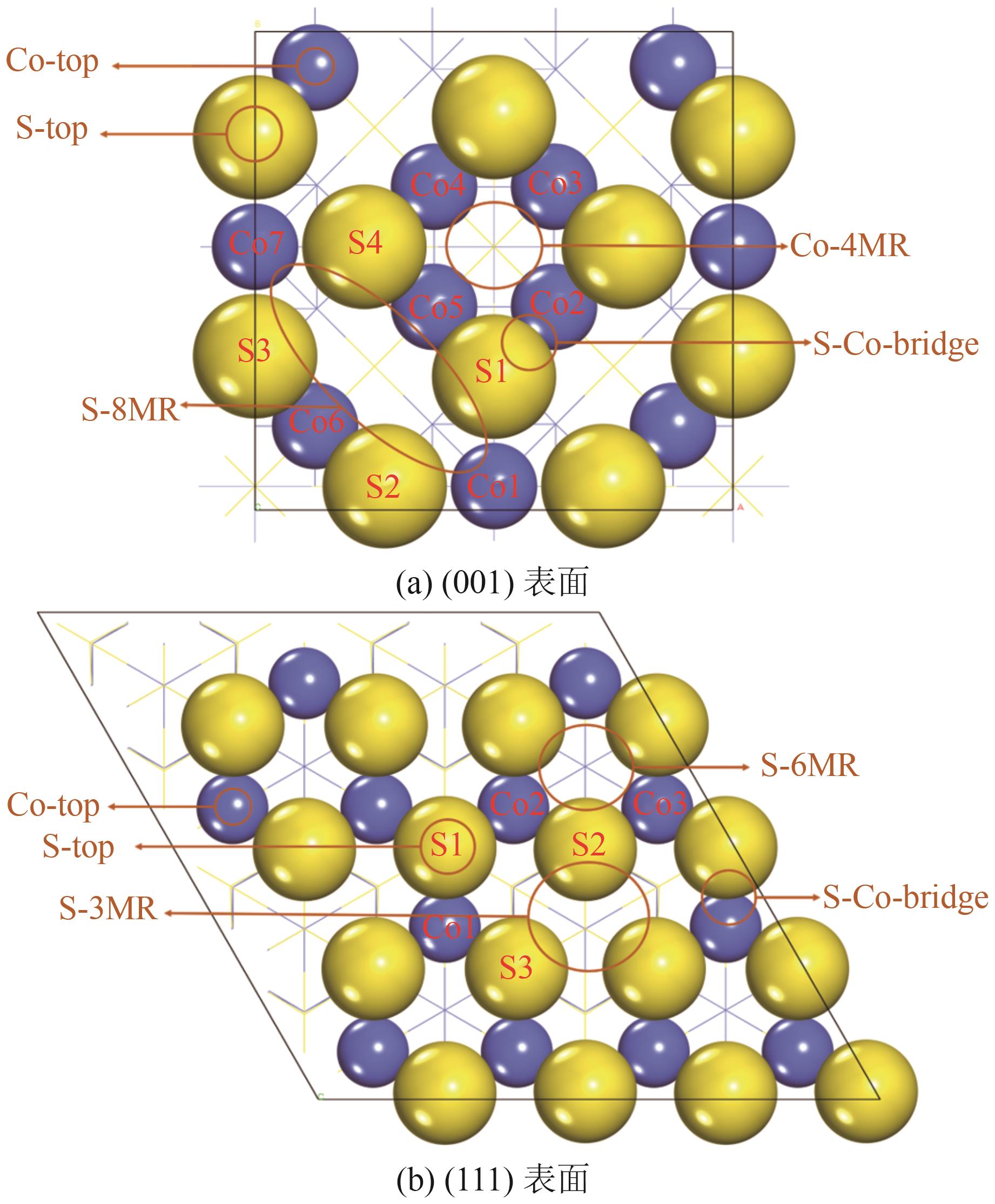
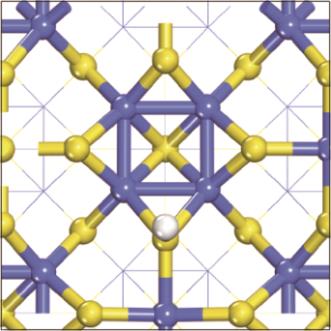
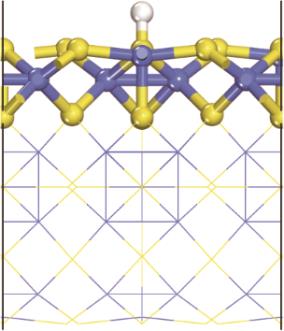
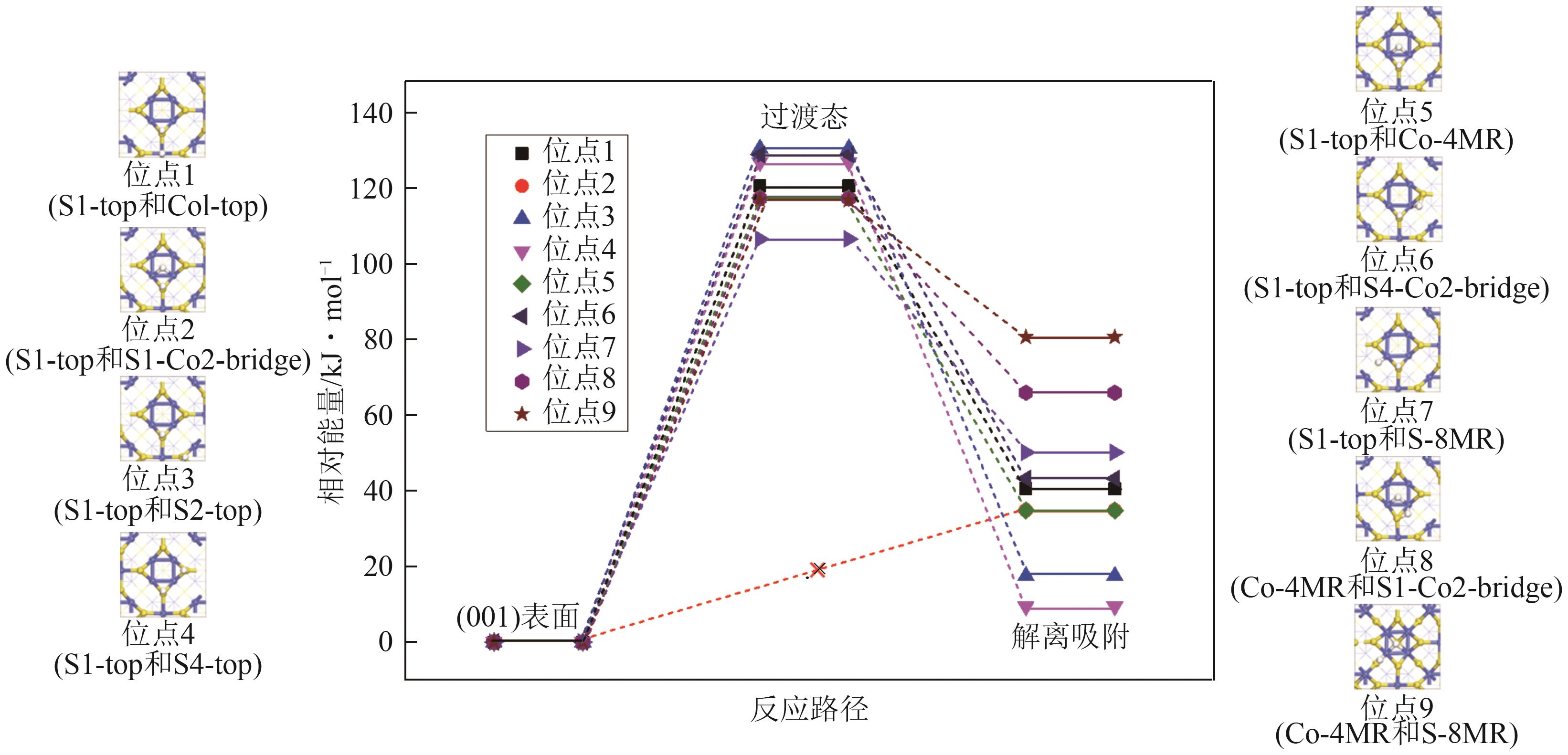
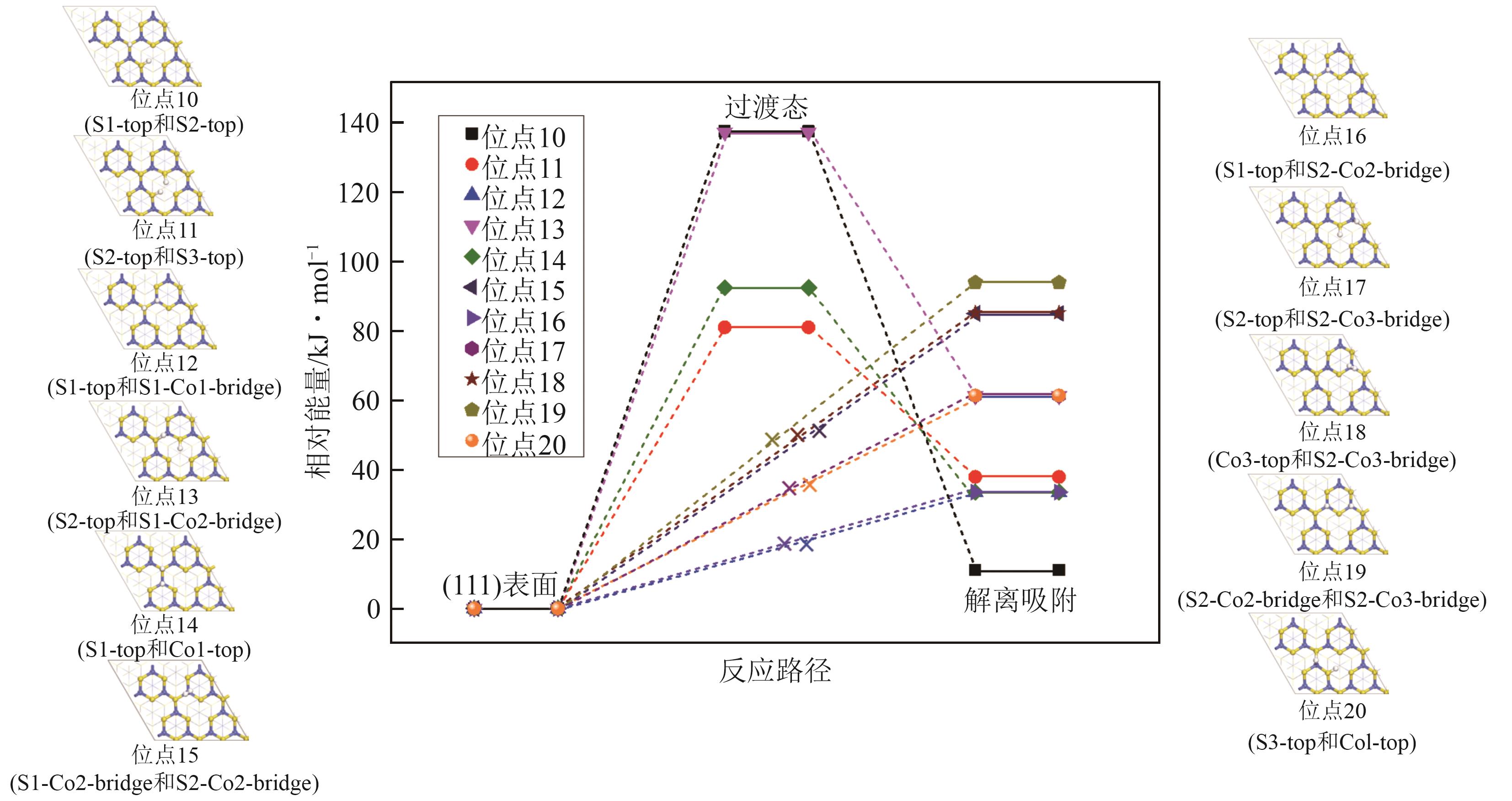
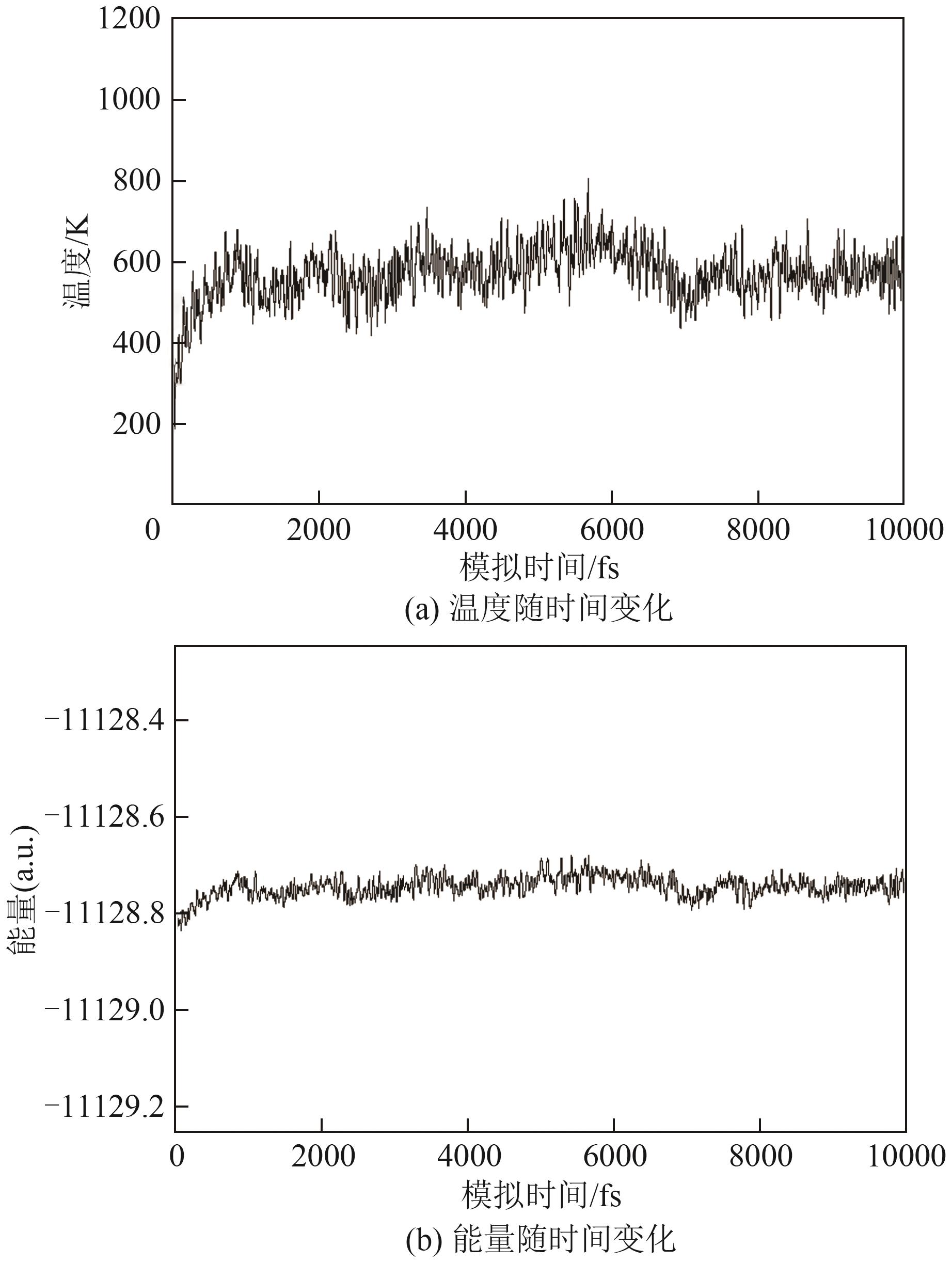
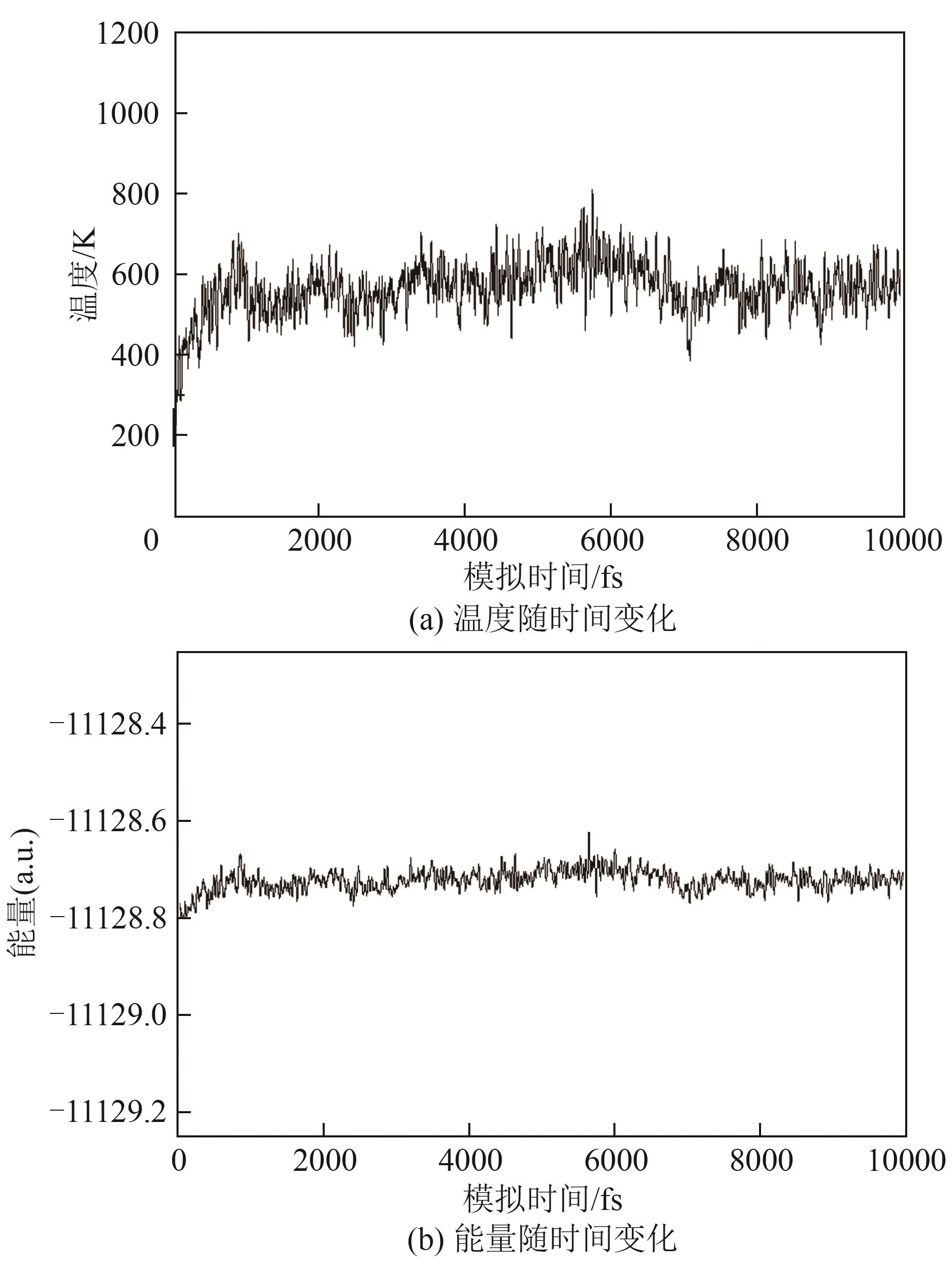
| [1] |
黄文斌, 魏强, 周亚松. 均一介孔Al2O3劣质蜡油加氢脱氮催化剂研究进展[J]. 化工进展, 2020, 39(S2): 196-203.
|
|
HUANG Wenbin, WEI Qiang, ZHOU Yasong. Research progress of homogeneous mesoporous Al2O3 of hydrodenitrogenation catalyst for inferior gas oil[J]. Chemical Industry and Engineering Progress, 2020, 39(S2): 196-203.
|
| [2] |
霍佳宁, 贾燕子, 胡大为, 等. 渣油加氢脱硫催化剂活性相结构调控研究进展[J]. 石油炼制与化工, 2024, 55(12): 147-154.
|
|
HUO Jianing, JIA Yanzi, HU Dawei, et al. Research progress in active phase structure control of residue hydrodesulfurization catalyst[J]. Petroleum Processing and Petrochemicals, 2024, 55(12): 147-154.
|
| [3] |
于沛, 柯明, 王奇, 等. Ce改性CoMo/Al2O3选择性加氢脱硫催化剂的表征及其催化硫醇硫生成性能[J]. 石油学报(石油加工), 2020, 36(5): 909-918.
|
|
YU Pei, KE Ming, WANG Qi, et al. Characterization and catalytic performance of Ce modified CoMo/Al2O3 catalyst for mercaptan sulfur formation in selective hydrodesulfurization[J]. Acta Petrolei Sinica (Petroleum Processing Section), 2020, 36(5): 909-918.
|
| [4] |
王薇, 赵晓光, 李会峰, 等. 噻吩在钴钼硫活性中心上的吸附及反应化学研究[J]. 计算机与应用化学, 2018, 35(4): 269-276.
|
|
WANG Wei, ZHAO Xiaoguang, LI Huifeng, et al. Adsorption and reaction of thiophene on CoMoS active sites[J]. Computers and Applied Chemistry, 2018, 35(4): 269-276.
|
| [5] |
孙进, 郭蓉, 陈晓贞, 等. 助剂Co对加氢处理催化剂性能的影响[J]. 石油炼制与化工, 2023, 54(6): 32-38.
|
|
SUN Jin, GUO Rong, CHEN Xiaozhen, et al. Effect of promoter cobalt on the performance of hydrotreating catalysts[J]. Petroleum Processing and Petrochemicals, 2023, 54(6): 32-38.
|
| [6] |
李宇航, 李若愚, 田丰宇, 等. Mo修饰NiS x /γ-Al2O3-TiO2催化剂的制备及用于柴油超深度脱硫反应性能[J]. 化工进展, 2025, 44(11): 6359-6367.
|
|
LI Yuhang, LI Ruoyu, TIAN Fengyu, et al. Performance of molybdenum modified NiS x /γ-Al2O3-TiO2 catalyst in ultra-deep diesel desulfurization[J]. Chemical Industry and Engineering Progress, 2025, 44(11): 6359-6367.
|
| [7] |
DIAO Xinyong, JI Na, LI Xinxin, et al. Fabricating high temperature stable Mo-Co9S8/Al2O3 catalyst for selective hydrodeoxygenation of lignin to arenes[J]. Applied Catalysis B: Environmental, 2022, 305: 121067.
|
| [8] |
XIAO Tao, WU Kui, WANG Dan, et al. Preparation of Co-Co9S8-MoS2 catalyst for efficient deoxygenation of lignin-derived aromatic oxy-compounds into arenes[J]. Fuel, 2024, 357: 129669.
|
| [9] |
RAMOS Manuel, BERHAULT Gilles, FERRER Domingo A, et al. HRTEM and molecular modeling of the MoS2-Co9S8 interface: Understanding the promotion effect in bulk HDS catalysts[J]. Catalysis Science & Technology, 2012, 2(1): 164-178.
|
| [10] |
HADJ-AÏSSA A, DASSENOY F, GEANTET C, et al. Solution synthesis of core-shell Co9S8@MoS2 catalysts[J]. Catalysis Science & Technology, 2016, 6(13): 4901-4909.
|
| [11] |
GRIMME Stefan. Semiempirical GGA-type density functional constructed with a long-range dispersion correction[J]. Journal of Computational Chemistry, 2006, 27(15): 1787-1799.
|
| [12] |
LU Tian, CHEN Feiwu. Multiwfn: A multifunctional wavefunction analyzer[J]. Journal of Computational Chemistry, 2012, 33(5): 580-592.
|
| [13] |
HENKELMAN Graeme, UBERUAGA Blas P, Hannes JÓNSSON. A climbing image nudged elastic band method for finding saddle points and minimum energy paths[J]. The Journal of Chemical Physics, 2000, 113(22): 9901-9904.
|
| [14] |
HENKELMAN Graeme, Hannes JÓNSSON. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives[J]. The Journal of Chemical Physics, 1999, 111(15): 7010-7022.
|
| [15] |
BUSSI Giovanni, DONADIO Davide, PARRINELLO Michele. Canonical sampling through velocity rescaling[J]. The Journal of Chemical Physics, 2007, 126(1): 014101.
|
| [16] |
REN Zhibo, LIU Ning, CHEN Biaohua, et al. Theoretical investigation of the structural stabilities of ceria surfaces and supported metal nanocluster in vapor and aqueous phases[J]. The Journal of Physical Chemistry C, 2018, 122(9): 4828-4840.
|
| [17] |
GELLER S. Refinement of the crystal structure of Co9S8 [J]. Acta Crystallographica, 1962, 15(12): 1195-1198.
|
| [18] |
GONZALEZ Gabriel A, ALVARADO Manuel, RAMOS Manuel A, et al. Stacking height effect and hydrogen activation calculations on the Co9S8/MoS2 catalyst via computational transition states[J]. Computational Materials Science, 2016, 123: 93-105.
|
| [19] |
ZHENG Peng, XIAO Chengkun, SONG Shaotong, et al. DFT insights into the hydrodenitrogenation mechanism of quinoline catalyzed by different Ni-promoted MoS2 edge sites: Effect of the active phase morphology[J]. Journal of Hazardous Materials, 2021, 411: 125127.
|
| [20] |
ZHANG Qi, SHANG Hui, XUE Zonghao, et al. The effect of microwave electric field on sulfur vacancies formation over the edge sites of Co/Ni-promoted and unpromoted MoS2 catalysts through DFT investigations[J]. Fuel, 2022, 318: 123553.
|
 ), HE Hangyu1, WANG Ruohan1, HU Lijun1, ZHANG Xiaohui1, YAN Xuemin1,2
), HE Hangyu1, WANG Ruohan1, HU Lijun1, ZHANG Xiaohui1, YAN Xuemin1,2